用户手册
>
软件使用
>
软件应用案例
>
LAMMPS
LAMMPS 是大规模经典分子动力学代码,代表大规模原子/分子大规模并行模拟器。LAMMPS 具有用于软材料(生物分子、聚合物)、固态材料(金属、半导体)和粗粒度或细观系统的潜力。它可用于对原子进行建模,或者更一般地说,它可用作原子、中观或连续介质尺度上的平行粒子模拟器。 支持模板提交和命令行提交两种作业运行方式,具体方法如下:
您可以通过如下几个入口进行模板提交作业
1)入口一:控制台页面点击作业提交,选择Lammps软件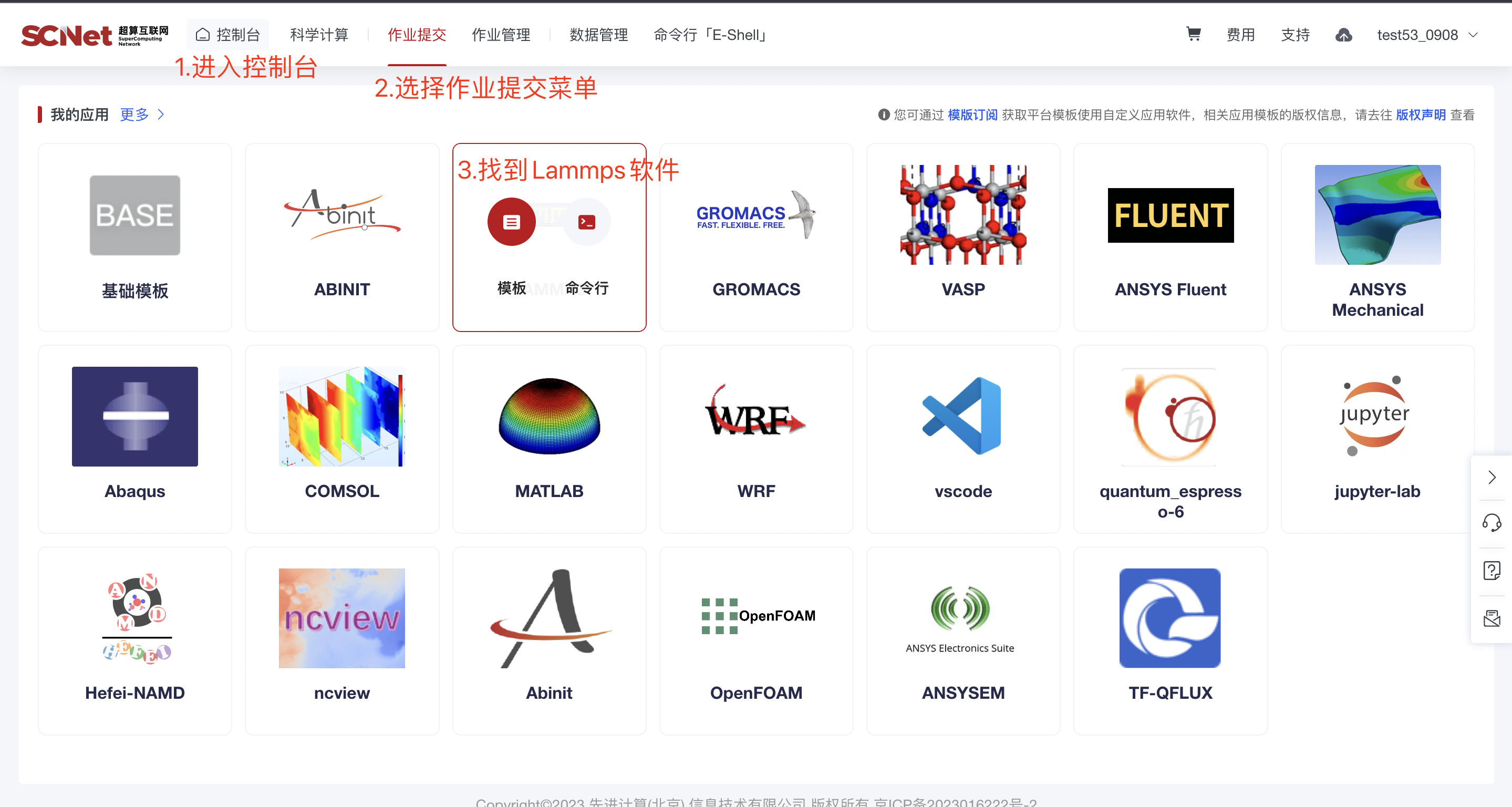 2)入口二:我的商品-应用软件菜单,选择Lammps软件
2)入口二:我的商品-应用软件菜单,选择Lammps软件 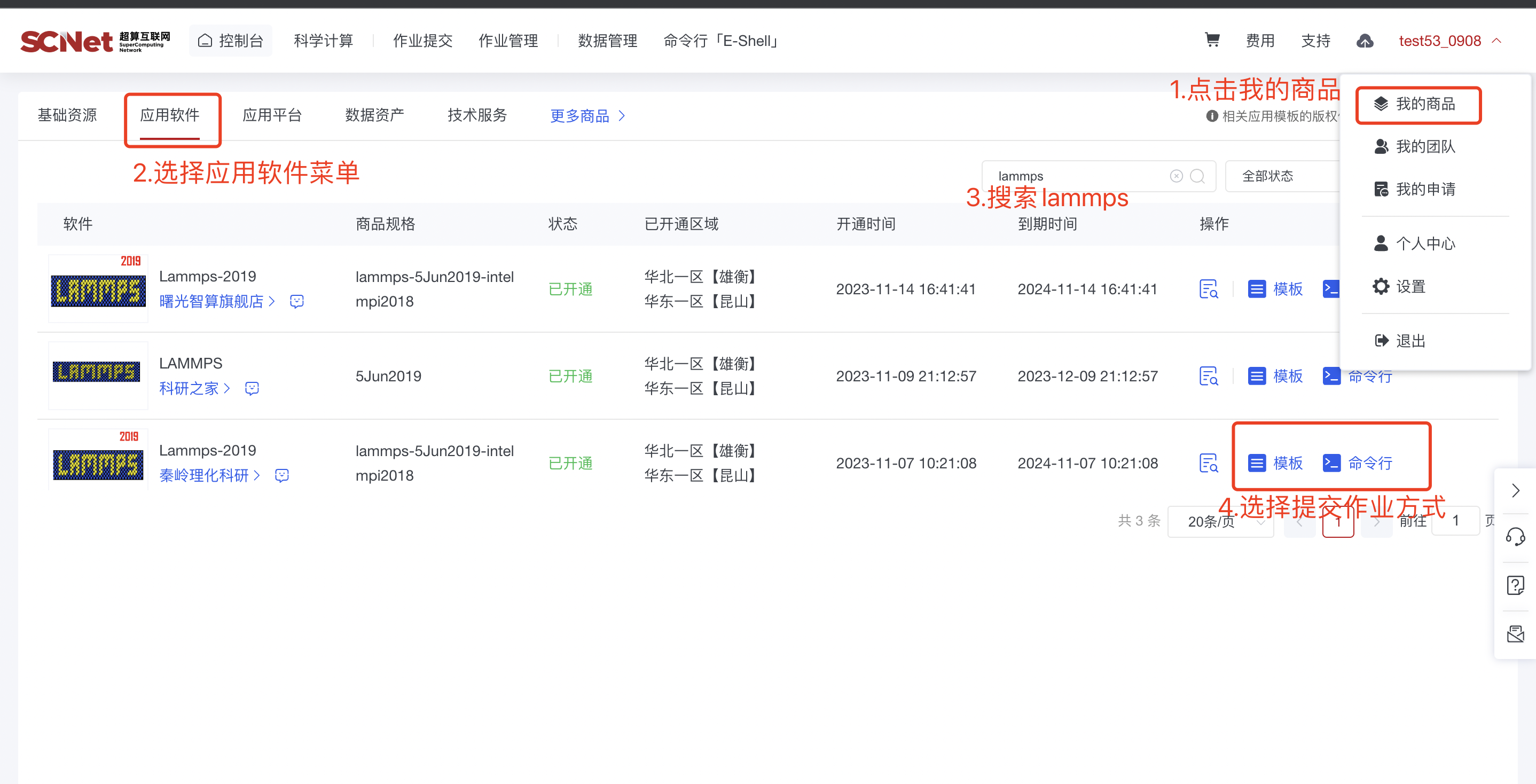
选择已开通区域对应的资源,填写相应的配置信息后,提交模版作业
【注意】根据任务申请的核数分配内存,核心/节点表示单节点使用的核数,最多不超过单节点总核数,可以点击队列下方的详情查看单节点配置的总核数。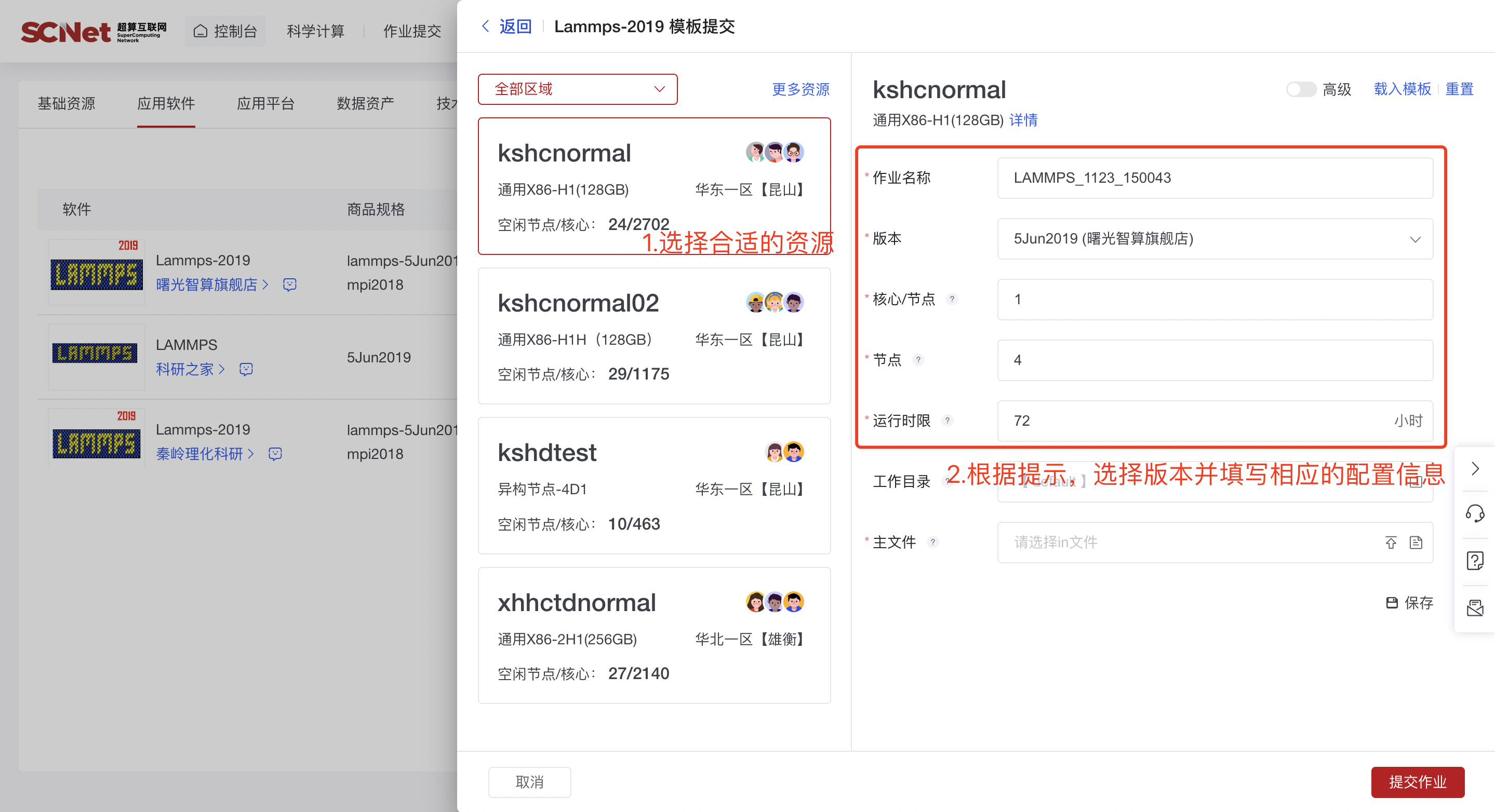 1)选择合适的计算资源后,再通过本地上传或者文件选择器选择文件,此处填写参数需要注意以下几点:
1)选择合适的计算资源后,再通过本地上传或者文件选择器选择文件,此处填写参数需要注意以下几点:
a.作业名称一般为默认,可按需求修改;
b.版本为安装软件的可执行程序路径;
c.核心/节点为每节点核数,与节点数相乘即为作业使用总核数;
d.作业时限为作业运行最大时长,作业超时即自动退出(即将超时但还在运行中的任务可以进行延时操作:在作业管理找到对应的作业号,点击作业号,在运行时限处点击延长来延长任务运行时间); e.工作目录默认为输入文件所在目录;
2)点击提交作业 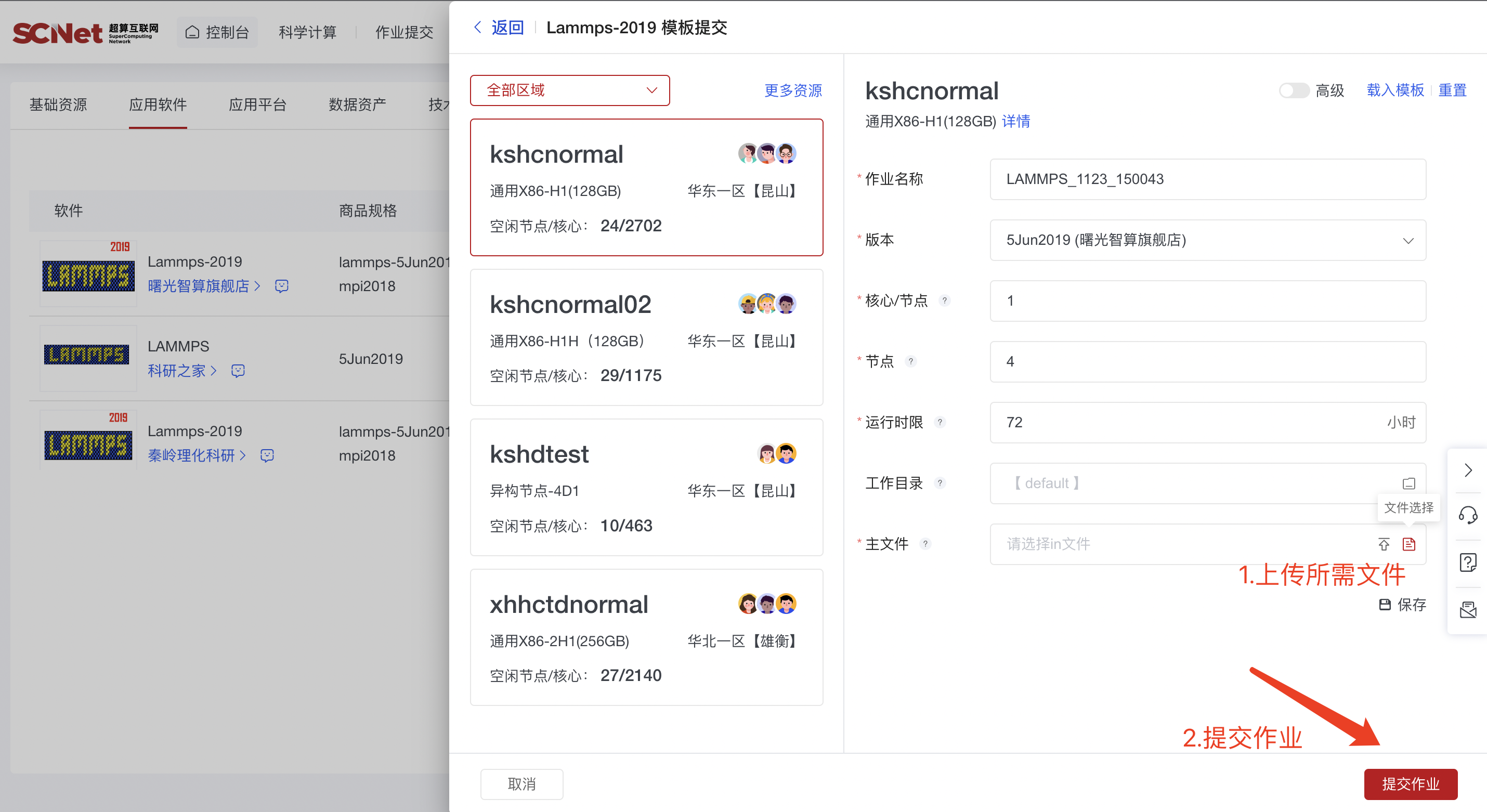
作业提交成功后,查看作业详情
1)模版提交作业后可查看运行的作业详情 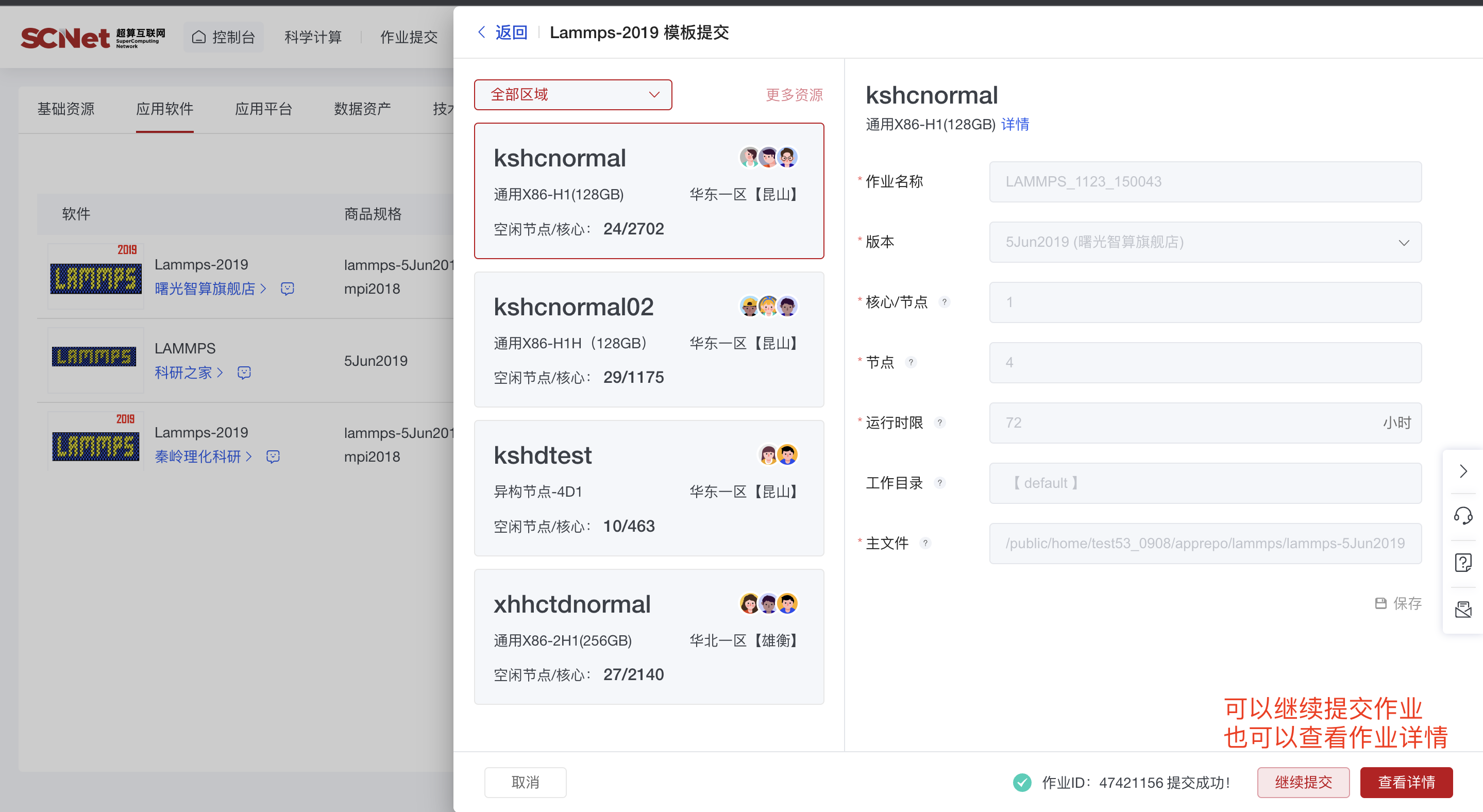 2)也可以通过作业管理入口,查看当前作业和历史作业
2)也可以通过作业管理入口,查看当前作业和历史作业 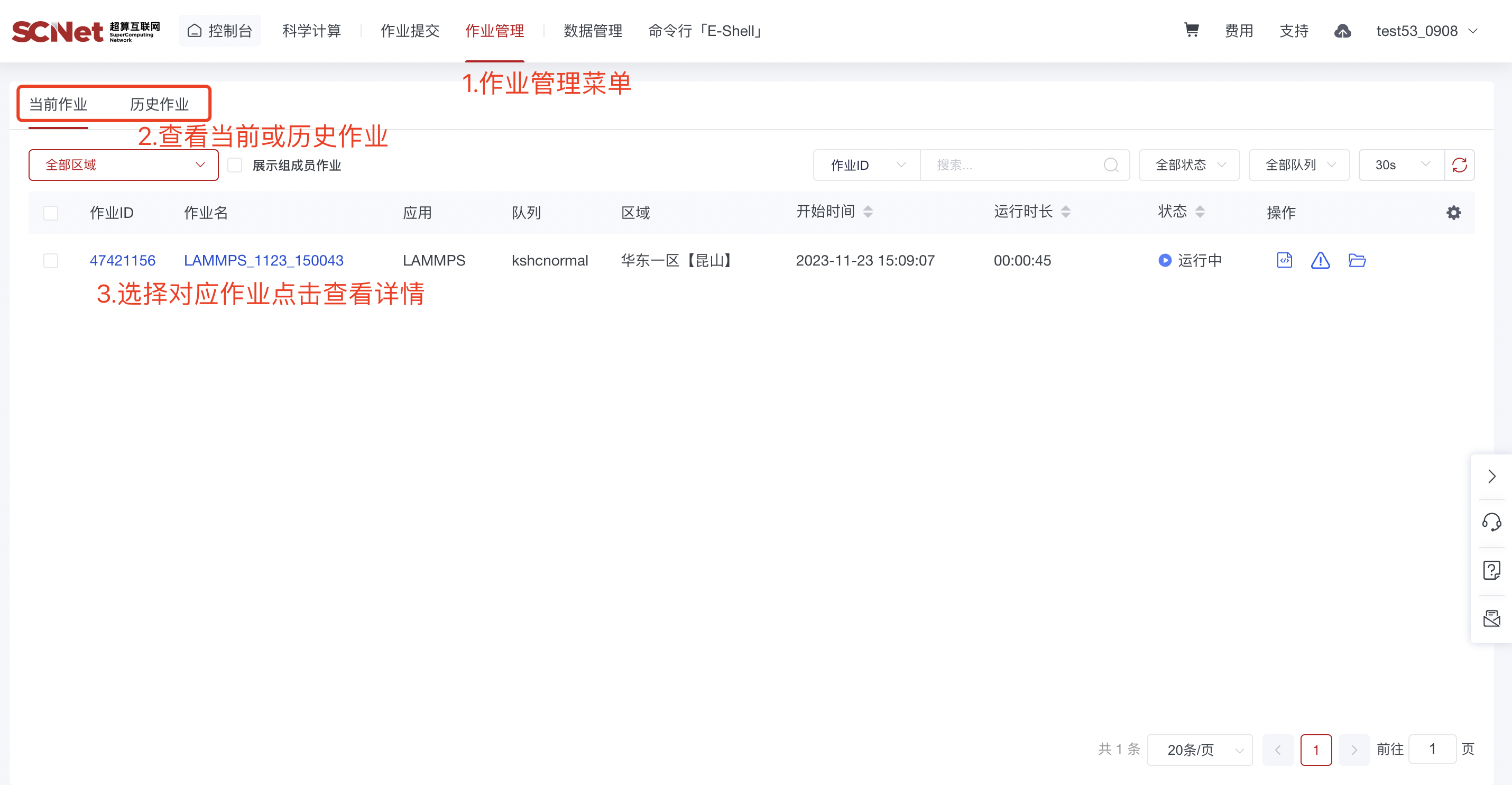 点击作业ID,查看具体的作业详情
点击作业ID,查看具体的作业详情 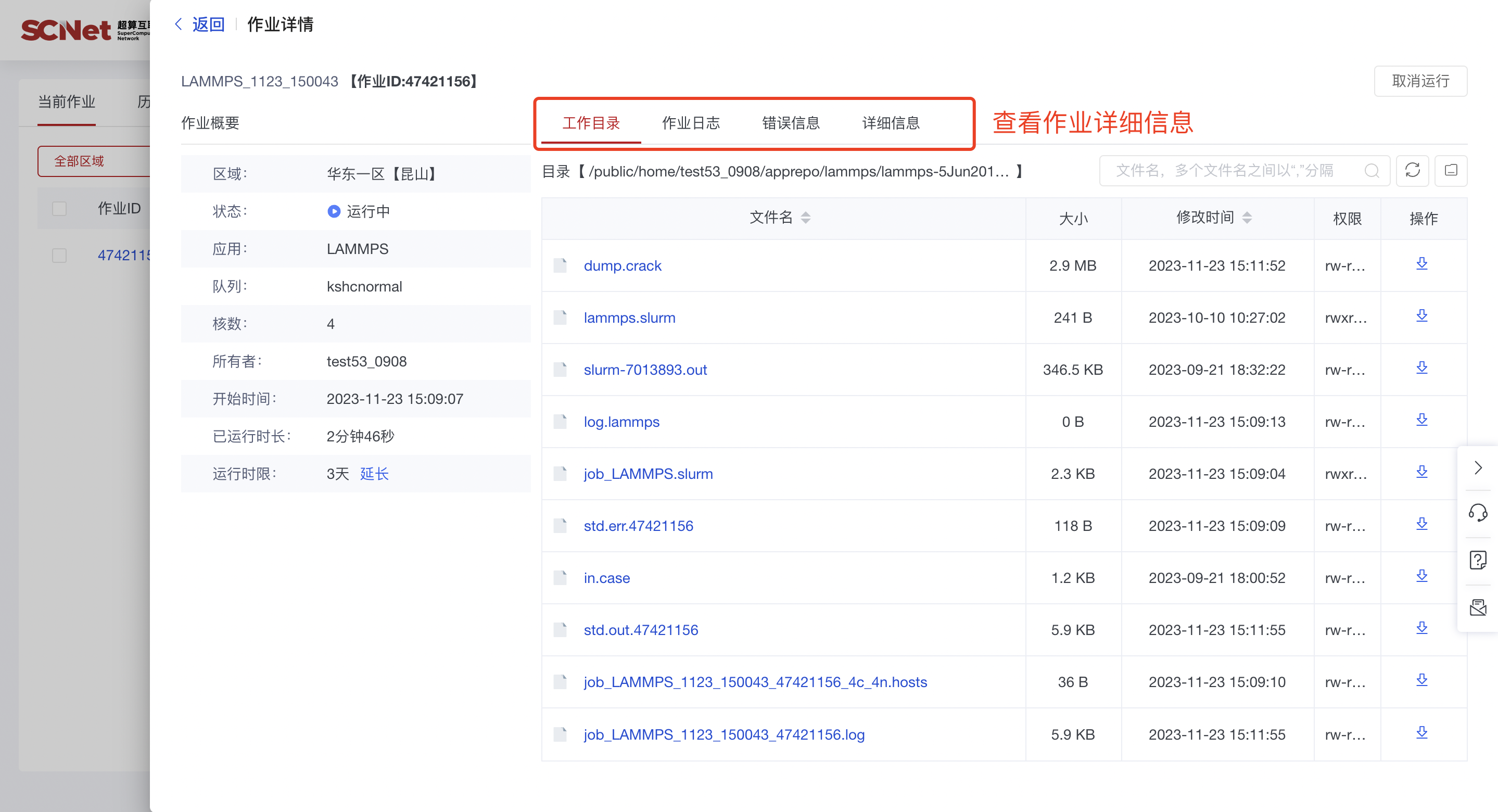

2)入口二:我的商品-应用软件菜单,选择Lammps软件 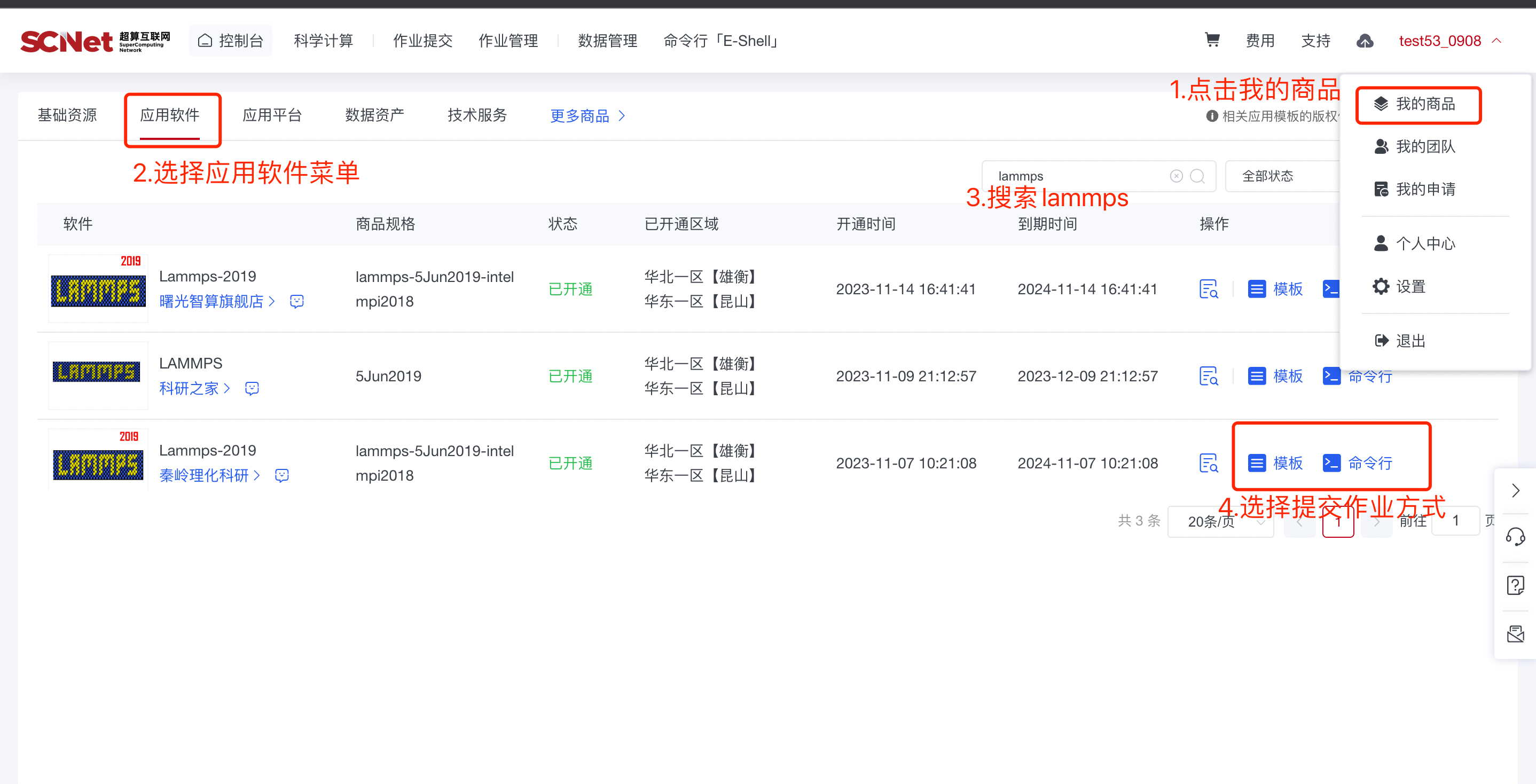
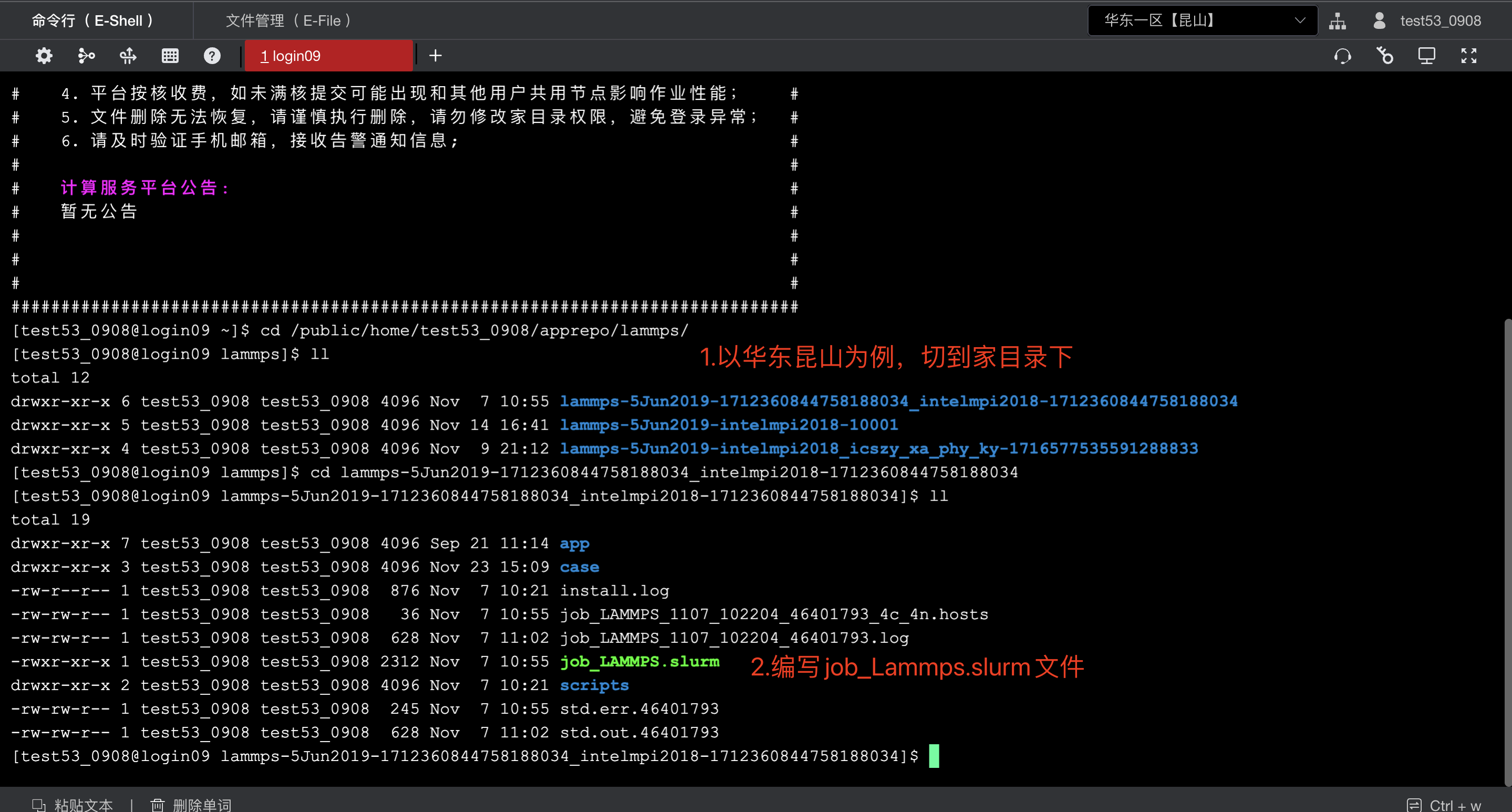
如下是一个参考文件
#!/bin/bash
#SBATCH -J LAMMPS_1107_102204 #作业名称
#SBATCH -p kshcnormal #队列
#SBATCH -N 4 #计算节点数
#SBATCH --ntasks-per-node=32 #每节点进程数
#SBATCH --time 72:00:00
#SBATCH --comment=LAMMPS
#SBATCH -o std.out.%j
#SBATCH -e std.err.%j
#Run LAMMPS
if [[ -z $NGPU ]] || [[ $NGPU -eq0]];then
OPT_GPU="" ; fi
if [[ -n $NGPU ]] && [[ $NGPU -ge1]];then
OPT_GPU="-sf gpu -pk gpu $NGPU" ; fi
cd $WORK_DIR
#module load compiler/intel/intel-
compiler2017.5.239mpi/intelmpi/2017.4.239
module purge
echo $MODULE_ENV > $MIDFILE_DIR/app.env
sed -i 's/_USER_COLON_SIGN_IN_GV_/:/g'
$MIDFILE_DIR/app.env
source $MIDFILE_DIR/app.env
mpirun $APP $OPT_GPU -in $IN_INPUT_FILE
2>&1 | tee -a $LOG_FILE
echo The end time is: `date +"%Y-%m-%d
%H:%M:%S"` | tee -a $LOG_FILE通过sbatch命令提交作业

通过squeue 命令查看作业运行情况 squeue
查看作业运行情况及参数详细介绍请点击查看SLURM命令。
结果文件下载请查看数据传输。