高性能计算服务
>
>
Quantum ESPRESSO(简称QE)是一套基于密度泛函理论、平面波和赝势开发的集成开源软件包,用于纳米级的电子结构计算和材料建模,它能够进行多种类型的计算,主要包括PWscf、Phonon等:
PWscf:执行平面波基组的自洽场(SCF)计算,可执行程序为pw.x等;
Phonon:计算声子谱和晶格动力学性质,可执行程序为ph.x等。
CP: 执行从头算(ab initio)分子动力学模拟,可执行程序为cp.x等。
在Quantum ESPRESSO中,pw.x模块是进行结构优化计算的核心工具,可以做自洽场(Self-Consistent Field, SCF)、结构优化(Structure Optimization)、能带结构(bands)、分子动力学(md)等计算。
本文,我们将带您深入浅出了解QE中的结构优化,阐述结构优化与自洽场(SCF)的关系,并展示如何在超算互联网使用QE的pw.x模块做结构优化计算。
结构优化是指通过调整原子的位置来找到体系的最低能量状态,这通常涉及到对原子坐标进行优化,使系统的总能量最小化。
听起来可能有些抽象,但实际上结构优化是材料科学和计算物理中一个至关重要的概念。简单来说,结构优化就是通过计算和调整,寻找一个分子或晶体结构的最低能量状态,即所谓的“全局能量最小化”。
如果不进行结构优化,可能会导致系统结构不稳定或者能量较高,无法准确描述实际体系的性质。因此,在实际应用中,通过结构优化可以帮助科研人员得到更为准确和可靠的结果,为后续的性质计算和研究提供基础。
结构优化与自洽场之间的关系紧密,是在材料科学和计算物理中不可或缺的步骤。
具体而言,SCF计算是量子力学中的一个基本步骤,用于求解多电子体系的基态波函数。在QE中,通过迭代方法求解Kohn-Sham方程,直到电子密度和波函数收敛到一定的精度。
通常做结构优化之前,需要先执行SCF计算,因为结构优化的目的是找到能量最低的原子配置,而这个能量计算是基于电子密度的。没有准确的电子密度,就无法正确计算原子间的作用力,也就无法进行有效的结构优化。而且,这个过程可能需要多次迭代SCF计算,因为每次原子位置的调整后,都需要重新进行SCF计算以更新电子密度和波函数。
可以简单理解为pw.x既能做SCF计算,也能做结构优化计算。做结构优化计算时,自洽场计算提供了结构优化所需的电子密度等信息,而结构优化则在此基础上进一步调整原子位置,以降低能量。通过这种迭代过程,科研人员可以获得材料的精确电子结构和结构状态。
第一步,准备QE软件和实验输入文件。如果您只想一心搞实验,不想花时间在编译和搭建环境上,超算互联网为您提供了多种版本、多种编译方式、链接库全面的Quantum ESPRESSO软件包,满足您即来即用的需求。
这里我们详细介绍基于超算互联网QE v7.0版本商品软件,如何购买商品一键启用该软件,以及如何进行结构优化计算和结果的可视化展示。
首先,登录超算互联网https://www.scnet.cn, 搜索“quantum espresso”,找到您所需的QE版本,选择计算资源(若无计算资源,需要先购买资源)、同意服务协议、去使用软件,等待软件部署完毕即可使用软件。
下面以做Ge原子结构优化的实验为例,详细介绍如何设置结构优化的输入文件。input文件如下,包含了一些主要的参数设置及其含义解释,后续命名为relax.in文件作为实验输入:
&CONTROL
calculation = "vc-relax" !设置计算类型为vc-relax
forc_conv_thr = 1.00000e-03 !力的收敛阈值
max_seconds = 1.72800e+05 !计算的最大时间
nstep = 100 !总步数限制
pseudo_dir = "./" !伪势文件目录
/
&SYSTEM
a = 4.04245e+00 !晶格参数,基面长度
c = 2.00000e+01 !晶格参数,垂直于基面的晶格长度
degauss = 1.00000e-02 !费米能级展宽
ecutrho = 2.37000e+02 !这两个设置电荷密度和波函数截断能
ecutwfc = 3.80000e+01
ibrav = 4 !晶格类型,4表示体心四方晶格
nat = 2 !原子数
ntyp = 1 !原子类型数
occupations = "smearing" !电子占据方式
smearing = "gaussian" !展宽类型
/
&ELECTRONS
conv_thr = 1.00000e-07 !电子结构的收敛阈值
electron_maxstep = 200 !电子迭代的最大步数
mixing_beta = 7.00000e-01 !混合参数,用于电子结构自洽场迭代
startingpot = "atomic" !这两个是初始势和波函数设置
startingwfc = "atomic+random"
/
&IONS !这里通常设置离子弛豫的参数
/
&CELL !这里通常设置晶格常数的优化参数
/
K_POINTS {automatic} !使用自动生成的k点网格
16 16 1 0 0 0
ATOMIC_SPECIES !指定了原子类型和对应的伪势文件。
Ge 72.61000 Ge.pz-dn-rrkjus_psl.0.2.2.UPF
ATOMIC_POSITIONS {angstrom} !指定原子的初始位置
Ge 1.010604 0.583468 10.366960
Ge 1.010621 2.917396 9.633040&CONTROl:包含了控制计算流程的参数,对于结构优化,calculation 参数通常设置为relax或者vc-relax,前者表示只允许原子位置变化,后者表示允许改变位置和晶格常数, nstep表示的是整个结构优化过程中离子弛豫的总步数限制,正如前面讲述的与SCF计算的区别,需要注意的是,ELECTRONS标签里还有一个步数设置electron_maxstep,表示的是电子自洽场(SCF)循环中,每次离子弛豫步骤中电子迭代的最大步数。
Tips: 离子弛豫(Ion Relaxation)和电子迭代(Electronic Iteration)是第一性原理计算中的两个重要概念。电子迭代,也称为自洽场(Self-Consistent Field, SCF)迭代,是指在固定原子位置的情况下,通过迭代求解Kohn-Sham方程来获得电子结构的过程。离子弛豫是指在给定的电子结构下,通过调整原子的位置来最小化系统的总能量的过程。这个过程通常在电子结构计算之后进行。
&SYSTEM部分用来定义系统的晶格参数和原子信息;
&ELECTRONS用来设置电子结构计算的参数比如收敛阈值、迭代步数等。
&IONS用于指定离子动力学相关的参数,主要用在结构优化计算中。以下是&ions部分中一些常用参数的具体解释:
ion_dynamics:指定离子动力学的类型(relax和vc-relax计算类型默认使用bfgs算法)
upscale:在结构优化过程中,conv_thr的最大减少因子,当优化过程中原子移动的步长较小时,可以适当增加这个值以加快收敛。
remove_rigid_rot:当设置为.true.时,Quantum ESPRESSO会尝试去除晶胞的刚体旋转,只优化晶胞的形状和大小。在模拟孤立系统的动力学或热力学时非常有用。
此外,实验还包含一些其他的选项和参数,如CONSTRAINS用来设置原子位置的约束条件,更多的关于pw.x功能的参数及设置,可以参考QE的官方文档:https://www.quantum-espresso.org/Doc/INPUT_PW.html
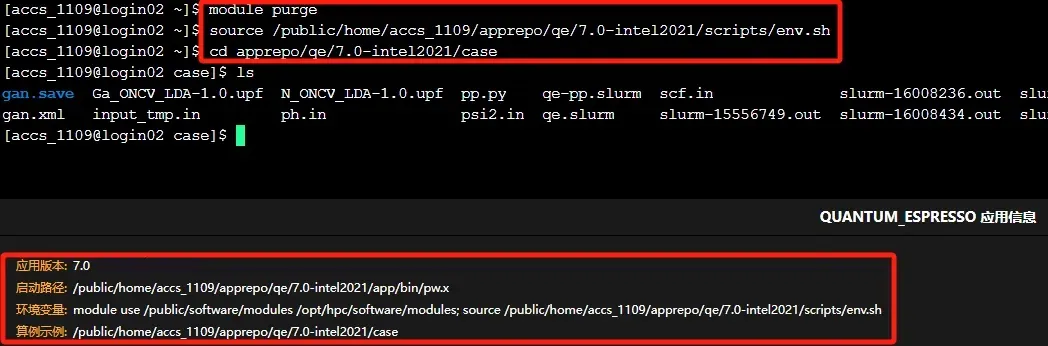
第二步,准备好输入文件以及相关的伪势文件(若需要)之后,就可以使用pw.x模块做计算了。您可在“超算互联网首页”个人账号中找到“我的商品”,选择“应用软件”标签,搜索找到购买的Quantum ESPRESSO软件,在右侧选择命令行使用软件。可根据命令行下方的提示信息加载环境变量和使用软件,软件默认安装在家目录的apprepo下,环境变量默认配置在软件包中scripts目录下的env.sh中。在软件case目录下,我们还放置了slurm脚本和算例案例,更多实践指南可参考QE商品详情页的使用手册。
pw命令和完整的qe.slurm脚本如下,执行sbatch qe.slurm即可提交作业计算。
##计算命令
mpirun -np <核心数> pw.x -i relax.in > relax.out
##slurm脚本
#!/bin/bash
#SBATCH -J qe-relax
#SBATCH -N 1
#SBATCH --ntasks-per-node=32
#SBATCH -p xahctar
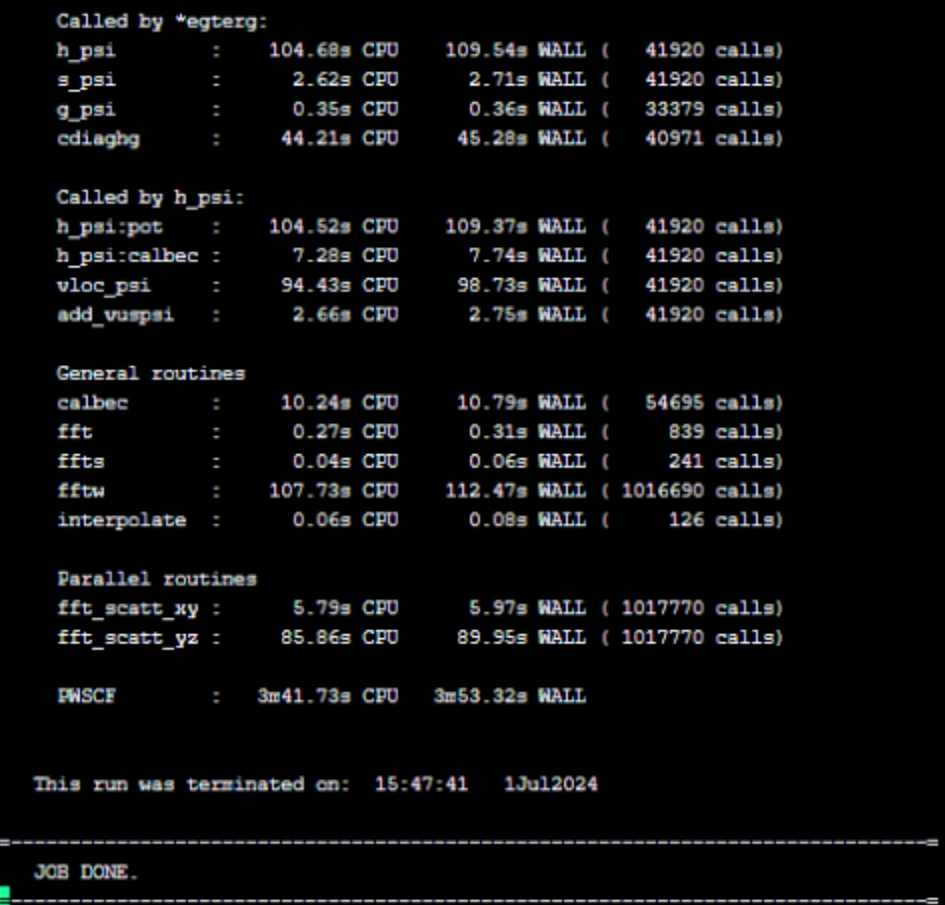
mpirun -np 32 pw.x -i relax.in > relax.out这里我们指定了输出重定向到relax.out文件,便于进行后处理操作。程序正常执行完成后,会有如下图所示的类似输出:
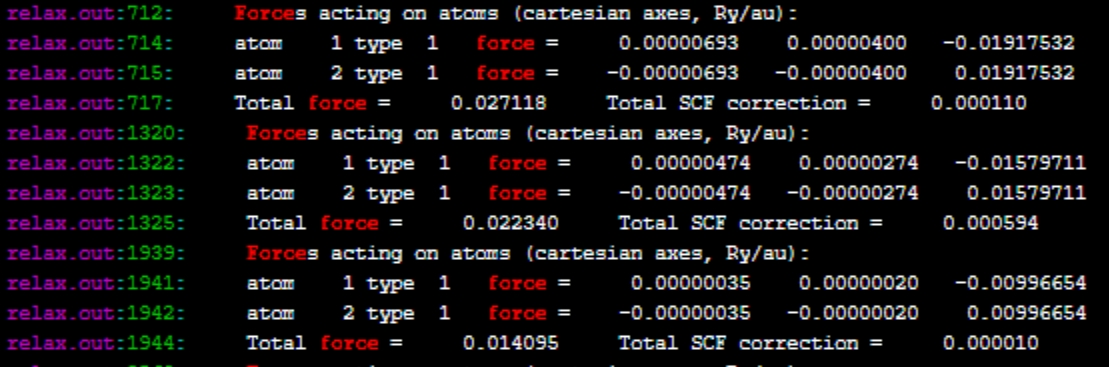
由于我们没有设置输出目录和文件名(可以在CONTROL中设置outdir、prefix参数),将会在算例目录下自动输出一个pwscf.save文件夹,里面有程序运行过程中保存的波函数数据,它包含了电子结构的详细信息。结构优化的计算过程输出保存在了relax.out,我们可以通过以下信息来识别能量和结构的变化,以及优化过程的细节:
优化步骤(Ion positions and cell parameters):输出通常会列出每次迭代中原子的位置和晶胞参数。通过比较连续迭代的输出,你可以观察到原子位置和晶胞参数的变化。
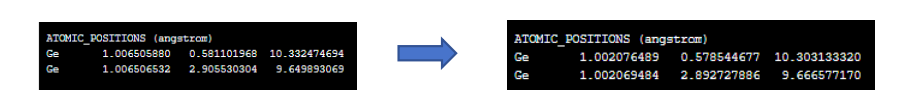
总能量(Total energy):每次迭代结束时,都会输出当前步的总能量(可使用grep命令直接搜索输出文件中的total energy)。

力(Forces):原子所受的力是结构优化的关键信息。如果输出中包含了每个原子的力的大小和方向,这表明进行了结构优化。力的收敛是优化过程的一个重要指标。
如上文通过查看文件做结构优化计算的方式,简直太不直观了!这里我们推荐使用QE的搭配软件XCrySDen来进行可视化展示结构优化过程。超算互联网提供XCrySDen 1.6.2版本,支持一键加载环境!购买使用方式可参考上文QE的使用流程。
由于是可视化软件,需要在使用前配置Linux图形桌面。您可在“超算互联网首页”选择右上角的“控制台”,在快捷入口或者图形桌面选项卡中点击“Liunx图形桌面”,选择使用软件对应的区域后,“打开桌面”即可打开一个图形桌面,记录您的显示端口号,在使用XCrySDen软件的命令行中添加export DISPLAY=<端口号>即可通过图形桌面显示XCrySDen软件。
命令行使用xcrysden软件,只需加载好环境变量,输入xcrysden即可在图形桌面处显示xcrysden的图形界面。选择“File”-->“Open PWscf”—>“Open PWscf Output File”,打开结构优化过程中的输出文件。也可以通过命令行的方式打开relax.out文件,然后进行后续操作,命令如下:
xcrysden --pwo relax.out打开文件后,选择“Display All Coordinates as Animation”,然后在Animation Control中调整图片展示形式、播放结构优化轨迹、保存成图片或者gif等。
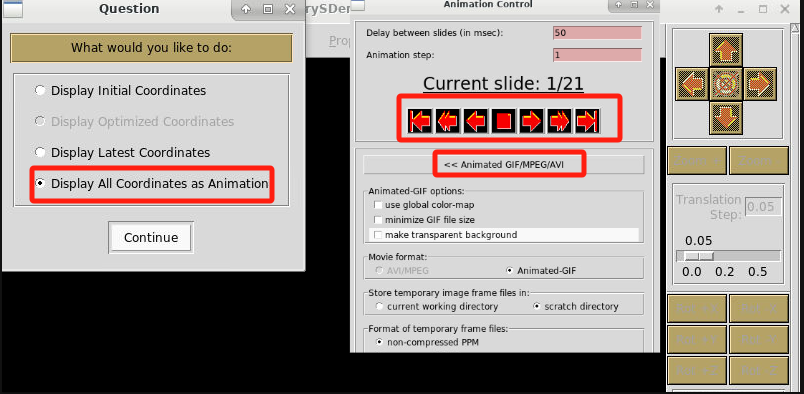
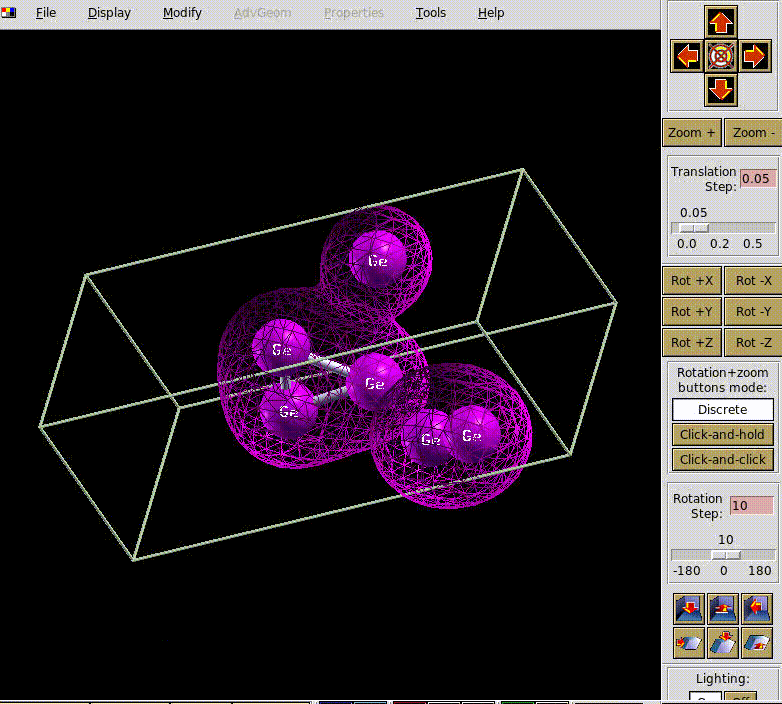
下面是我们测试算例的计算过程动态展示,这里为了效果明显,放置了6个Ge原子。