高性能计算服务
>
>
本篇为“超链接”系列专题的第3篇QE自洽计算最佳实践,您将获得pp.x电荷密度后处理与VESTA可视化展示。
Quantum ESPRESSO的自洽场(Self-Consistent Field,SCF)计算是一种计算物理方法,用于在密度泛函理论(Density Functional Theory, DFT)框架下求解多电子体系的基态性质。SCF计算主要进行以下内容:
1、电子密度的计算:通过迭代过程确定体系的基态电子密度。
2、波函数的优化:计算并优化电子的波函数,这是描述电子状态的量子力学函数。
3、总能量的计算:计算体系的总能量,包括动能、势能和交换-相关能。
本文将详细介绍如何在超算互联网使用Quantum ESPRESSO v7.2版本进行自洽计算实验,使用pp.x做后处理得到电荷密度图,并用VESTA软件进行可视化展示。
目前,超算互联网提供预编译的Quantum ESPRESSO 7.2版本,支持一键购买使用。本文实验测试使用intelmpi2021编译的QE 7.2版本:
您可以通过“我的商品”,在“应用软件”中找到或者搜索该版本商品,点击“命令行”,即可选择区域中心、命令行快速使用软件。
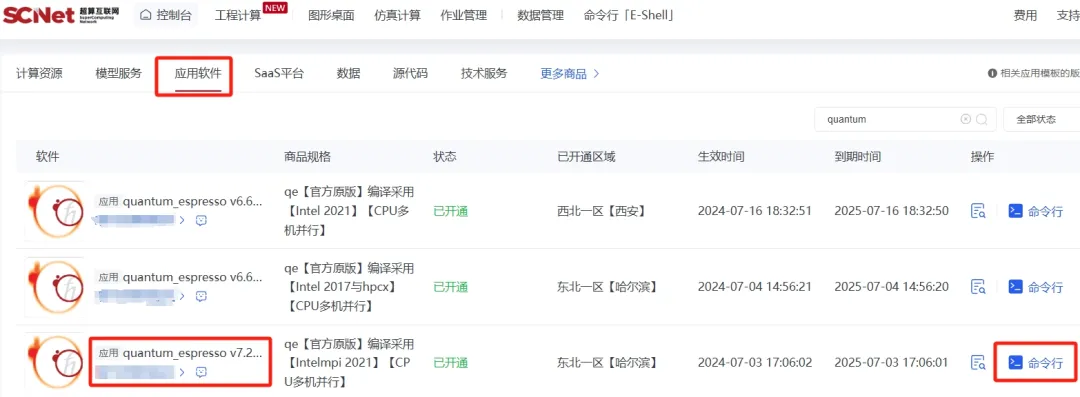
软件默认安装在家目录的apprepo下,环境变量默认放在软件scripts目录下的env.sh中,可执行如下命令添加环境变量:
source ~/apprepo/qe/7.2-intelmpi2021/scripts/env.sh此外,超算互联网还提供软件运行测试算例和slurm作业脚本,用户可以直接提交作业,运行算例。本次实践,我们使用软件case目录下的gan算例做scf计算,输入文件粘贴如下
&control !计算控制参数
calculation='scf' !表示自洽场计算
prefix='gan' !输出文件的前缀为gan
restart_mode='from_scratch' !重启模式设置为从头计算
pseudo_dir='./'
outdir='./'
tprnfor=.true. !打印力的选项为真
tstress=.true. !打印应力的选项为真
/
&system !该区域定义系统的结构信息
ibrav = 4 !表示体心四方晶格
celldm(1)=5.95484286816
celldm(3)=1.63011343669
nat = 4 !原子总数为4
ntyp = 2 !原子类型有2种(Ga和N)
ecutwfc = 160 !波函数截断能设置为160Ry
/
&electrons !定义电子计算参数
diagonalization='david' !对角化方法为Davidson
mixing_beta=0.7 !混合参数beta设置
conv_thr=1.0d-10 !电子迭代的收敛阈值
/
ATOMIC_SPECIES !定义了原子类型和对应的赝势文件
Ga 69.723 Ga_ONCV_LDA-1.0.upf
N 14.007 N_ONCV_LDA-1.0.upf
ATOMIC_POSITIONS crystal !以晶体坐标定义原子位置(Ga和N在晶格中的位置)
Ga 0.666666666667 0.333333333333 0.0
N 0.666666666667 0.333333333333 0.376429222
Ga 0.333333333333 0.666666666667 0.5
N 0.333333333333 0.666666666667 0.876429222
K_POINTS automatic !定义K点网格
6 6 4 0 0 0GaN是一种宽带隙半导体,广泛应用于电子和光电子领域。实验算例设置了体心四方晶格结构,具有两个原子类型(Ga和N),并指定了相应的赝势文件。直接使用case目录下的qe.slurm脚本即可提交作业,使用命令如下:
sbatch qe.slurm提交作业后,会提示作业id,使用该作业号,可以查询和跟踪作业情况,以下是两个常用的作业相关命令,其他命令和指导可参考slurm文档或商品对应的使用手册。
squeue #查看作业队列
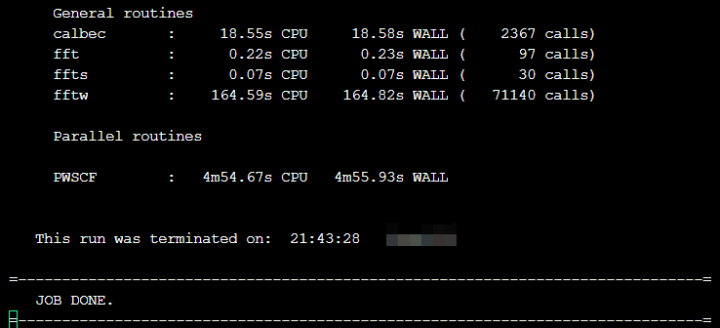
sacct –j <job-id> #查看作业运行情况等待作业完成后,会在当前目录生成以“gan”(输入文件设置的前缀)开头的文件夹,里面包含了运行结果输出文件,通过查看slurm-.out,可以看到类似如下程序的过程输出,“JOB DONE”表示程序正常计算结束。
pp.x 是 Quantum ESPRESSO 软件包中的一个程序,其主要目的是数据分析和绘图。该代码执行两个步骤:
1、读取 pw.x 生成的输出,提取并计算所需的量(电荷密度 rho,势能 V等)
2、将所需的量以适合各种绘图类型和绘图程序的格式写入文件
pp.x 提供了一种快捷的方式来分析和理解 pw.x 计算得到的数据,使得复杂的科学数据可以通过图形的方式直观展示,从而帮助研究人员更好地理解材料的电子结构和其他物理特性。若要使用pp.x做处理,需要按照一定的规则制定输入文件,下面是本算例的pp.x输入设置,这个 文件配置了从 pw.x 计算生成的电荷密度文件中提取电子(伪)电荷密度,并将其转换为 XSF 格式的文件 gan.rho.xsf,以便于后续的可视化分析:
&inputpp !包含了 pp.x 程序的主要输入参数
prefix = 'gan' !设置计算的前缀名称为 gan,这通常与 pw.x 计算使用的前缀相同。
outdir = './' !设置输出目录
filplot= 'gan.rho' !指定要处理和绘图的文件名为 gan.rho
plot_num = 0 !指定(选择)保存到filplot的类型
/
&plot !主要定义绘图的具体参数
filepp(1)= 'gan.rho' !要读取的文件为 gan.rho
iflag = 3 !指定绘图类型是3D plot
output_format = 5 !指定输出格式5,xcrysden 3D格式
fileout = 'gan.rho.xsf' !输出文件名关于pp.x输入文件的更多设置,可以参考QE官方文档指导,链接如下:https://www.quantum-espresso.org/Doc/INPUT_PP.html
将上述输入文件保存到pp.in文件,并放置在pw.x的输出文件gan.save同目录下,使slurm提交作业,即可运行后处理计算。slurm作业脚本如下:
#!/bin/bash
#SBATCH -J pp-test
#SBATCH -N 1
#SBATCH --ntasks-per-node=2
#SBATCH -p <队列名>
module purge
source /PATH/TO/YOUR/scripts/env.sh
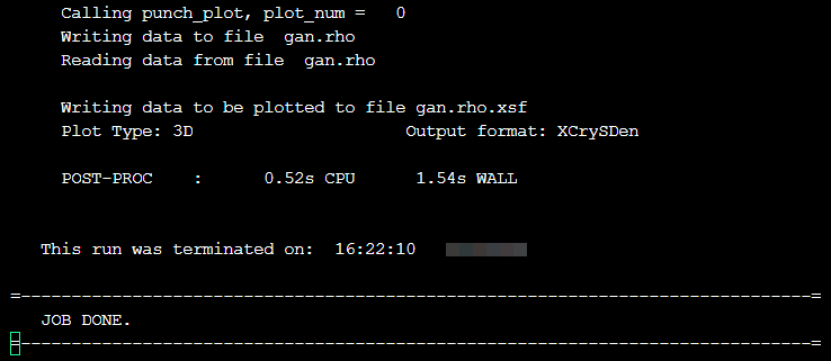
mpirun pp.x -in pp.in程序运行过程输出如下图所示,表示正常执行完毕,并且会在当前目录下生成gan.rho和gan.rho.xsf文件,“.xsf”文件可以用可视化软件绘图。
为了更直观显示,我们推荐用超算互联网的VESTA软件或XCrySDen软件绘制PP后处理得到的“xsf”文件,这里我们选择VESTA软件进行可视化展示,链接如下:

购买软件后,点击“去使用”,在命令行添加VESTA环境变量,设置图形桌面的DISPLAY端口号,即可在Linux图形桌面中使用软件。相关命令如下:
#加载VESTA环境变量
source ~/apprepo/VESTA/gtk3-none/scripts/env.sh
#添加图形桌面端口号export DISPLAY=YOUR PORT
#执行软件
VESTALinux图形桌面需要在使用软件前,通过“控制台”-->“图形桌面”-->“Linux图形桌面”,选择与VESTA软件相同的区域,开通后会显示对应的端口号。
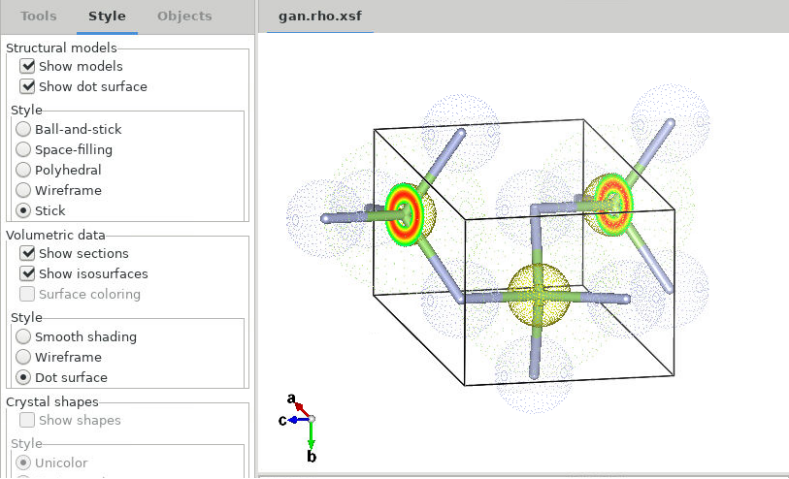
首先,打开VESTA软件,通过“File”-->“Open”找到并选择“gan.rho.xsf”文件,即可显示3D电荷密度图。您还可以自由选择“Tools”、“Style”和“Objects”选项显示风格和原子。例如通过调整左侧的“Tools”工具栏,选择“Animation”,可根据不同的轴心演示动画旋转效果。
下图展示了在“Style”选项中“Structural models”同时选择“Show models”和“show dot surface”,风格选择“Stick”;“Volumetric data”选择“show sections”和“Show isosurface”风格为“Dot surface”时的绘图效果。