高性能计算服务
>
>
VASPKIT是一个用于辅助 VASP(Vienna Ab-initio Simulation Package) 软件的工具包, 旨在帮助科学家和工程师更高效地进行第一性原理计算和材料模拟。VASPKIT 作为 VASP 的辅助工具,提供了丰富的功能和工具,包括输入文件生成、数据处理、结果分析等,可以帮助用户更便捷地进行 VASP 计算和后处理。现在,超算互联网上线 VASPKIT v1.2.5 版本,支持开箱即用。
本次实操,我们将以电催化氮还原反应理论计算为例,详细介绍如何在超算互联网使用 VASPKIT v1.2.5 版本对电催化反应第一性原理计算进行预处理和后处理。
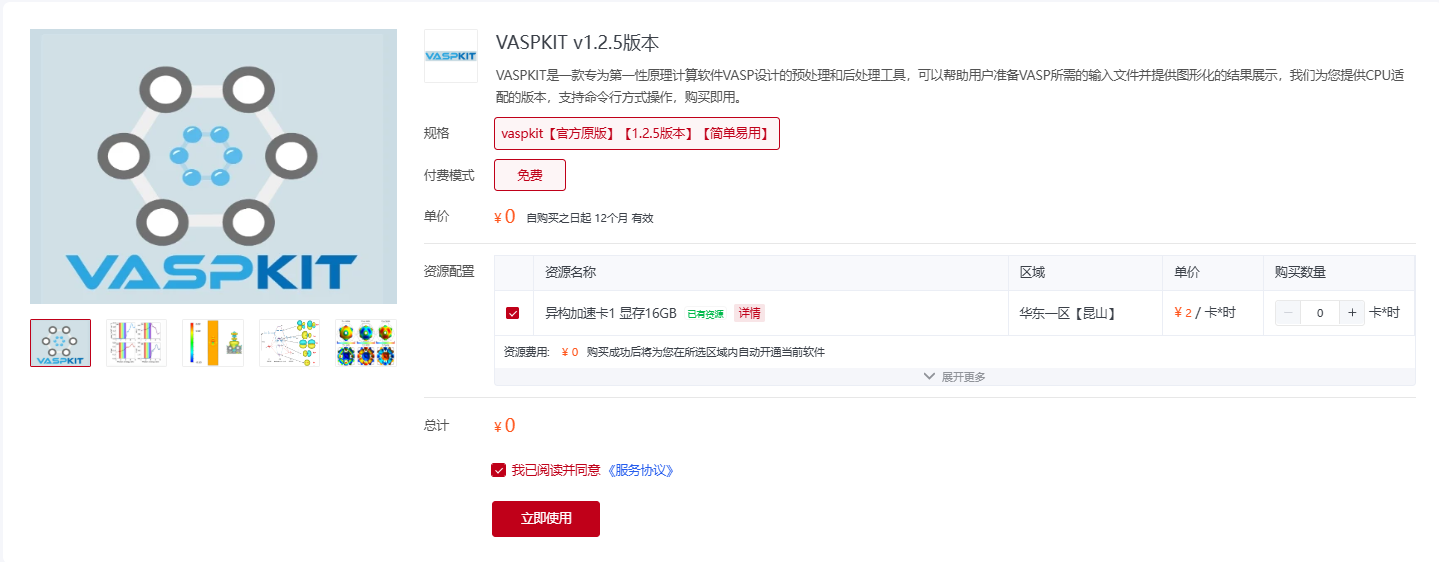
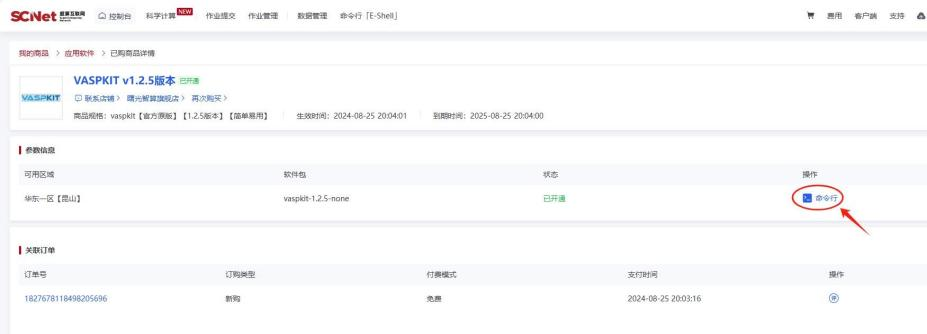
现在,超算互联网提供的 VASPKIT v1.2.5 版本,支持开箱即用。您可以访问下方链接,点击“立即使用”,进入“命令行”,即可快速使用 VASPKIT 软件。

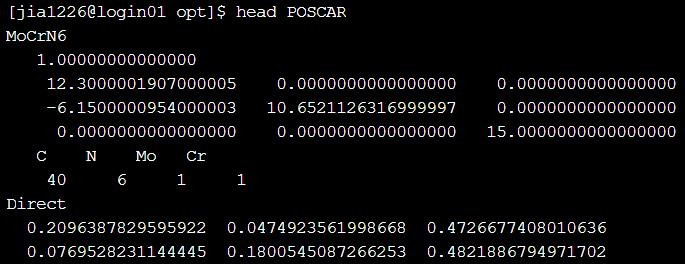
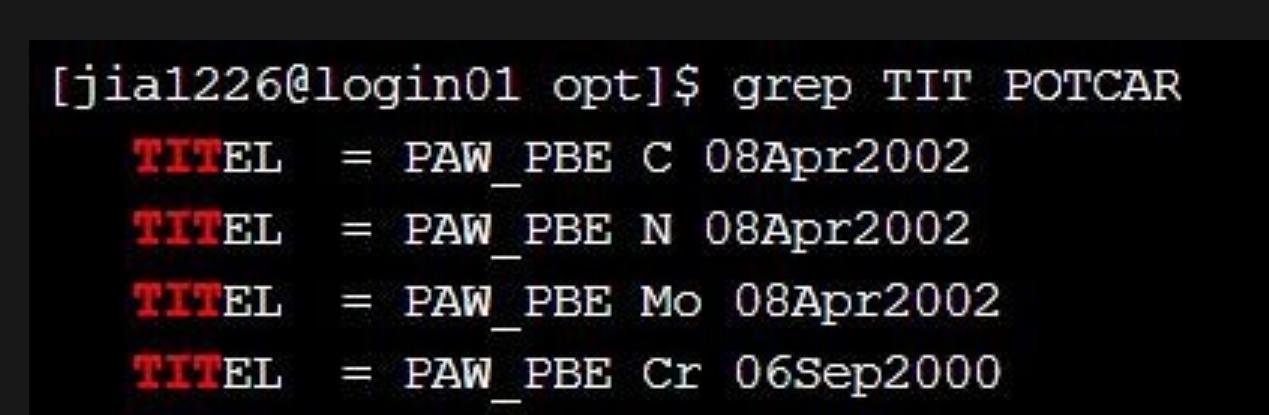
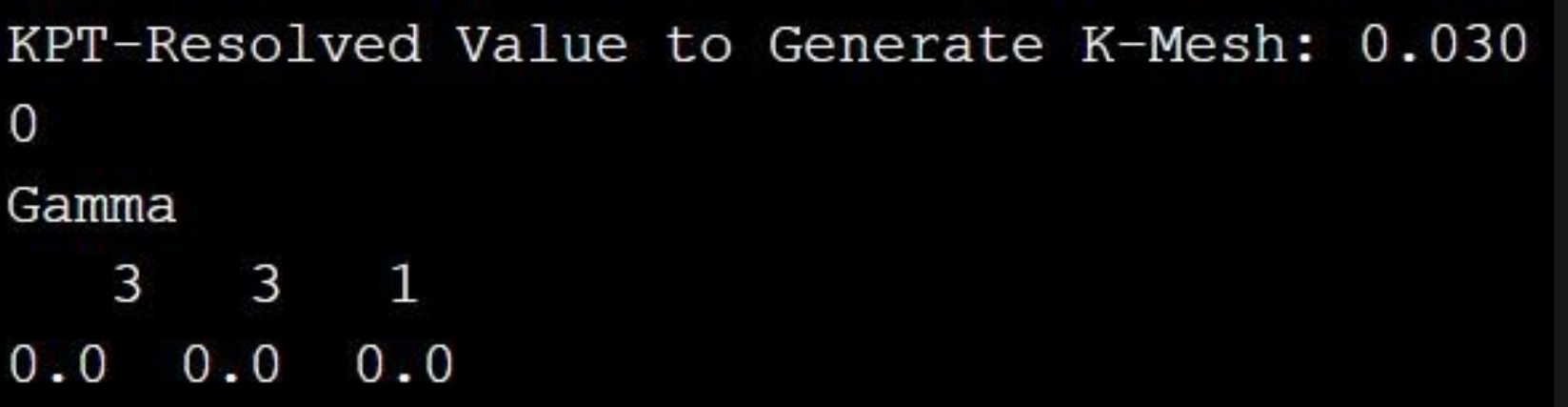
首先,我们使用软件 case 目录下的 NRR(氮还原反应)算例做 opt 计算,准备好 POSCAR(结构信息)、POTCAR(赝势信息)和 INCAR(计算参数信息)输入文件后,依此输入vaspkit-1-102-2-0.03,生成 VASP 计算所必须的 KPOINTS(K 点分布)文件,使用 vasp 提交脚本提交任务即可对结构进行优化。

优化完创建一个 scf 文件夹,对体系做静态计算,来得到后续电子结构计算所需要的CHGCAR(电荷密度文件)和 WAVECAR(波函数文件)等。注意这一步的 POSCAR 是由opt 计算中的 CONTCAR(优化后的稳定构型)复制而来的,POTCAR 和 KPOINTS 不变, 而 INCAR 需做以下修改:

创建一个 band 文件夹, 将静态计算得到的 CONTCAR 复制到 band 文件夹, 并修改为POSCAR,INCAR 在压缩包,将 CHGCAR 和 WAVECAR 以及 POTCAR 也复制进来,vaspkit-3-302,得到具有高对称点的 K 点路径,即 KPATH.in,复制成 KPOINTS 即可, 进行能带结构计算。
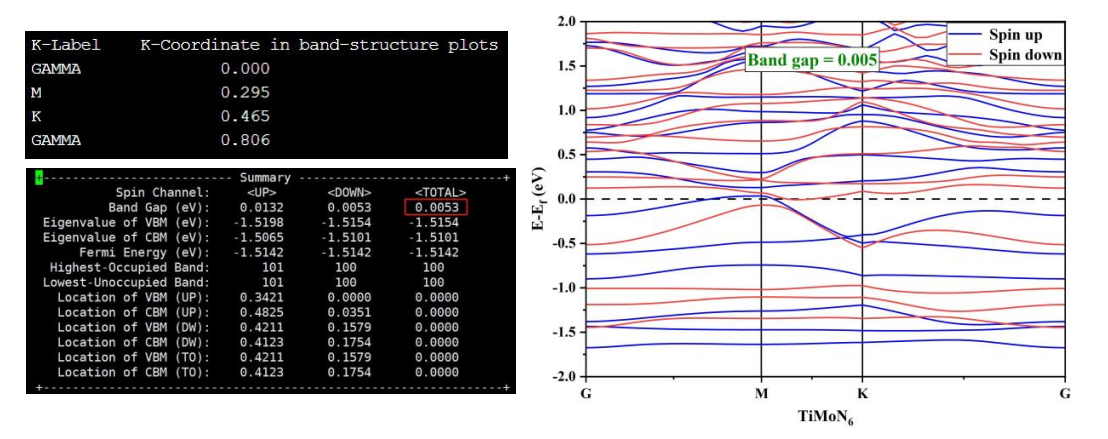
能带计算后处理:算完后 vaspkit-21-211,生成 BAND.dat,导出到 Origin 作图, 因为我们主要想看费米能级处的能带结构,所以我们可以将纵坐标调小一点( -2,2),而横坐标是由体系的高对称点信息决定的,图中的 G、M、K 是体系的高对称点,我们可以在 KLABELS 文件中查看此信息, 可以看到横坐标范围是( 0.000, 0.806)。打开 BAND_GAP(如下)可以看到体系的禁带宽度约为 0.0053 eV,低的能隙表明体系表现出金属性, 导电性较好, 利于电催化反应的进行。
创建一个 dos 文件夹, 将静态计算得到的 CONTCAR 复制为 POSCAR, 复制到dos 文件夹,INCAR 在压缩包,将 CHGCAR 和 POTCAR 也复制到 dos 文件夹,vaspkit生成 KPOINTS 文件, 进行态密度计算。注意计算 dos 时的计算精度应高于 opt 的, 得到的结果才更加准确, 所以我们要运行 vaspkit-1-102-2-0.01,增大 K 点网格。
态密度计算后处理: vaspkit-11-114-all;
vaspkit-11-115-Cr-5 6 7 8 9 这样就得到过渡金属元素的 d 轨道( dxy, dyz, dzx, dx2-y2 和 dz2)的投影态密度信息。输出文件是 PDOS_SUM.dat,导出文件用 Origin 打开作图;
vaspkit-11-115-N-2 3 4 得到 N 元素的 p 轨道( Px, Py 和 Pz) 的投影态密度图;
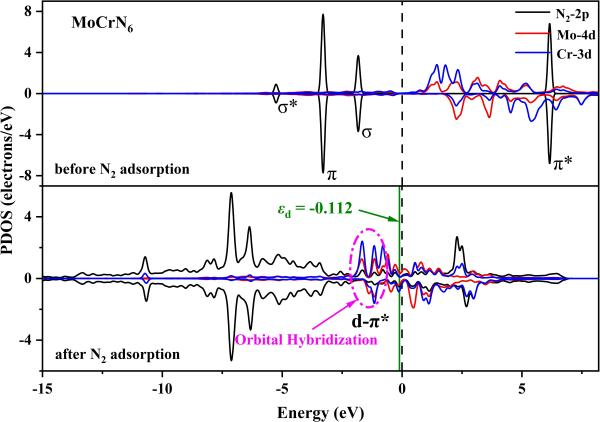
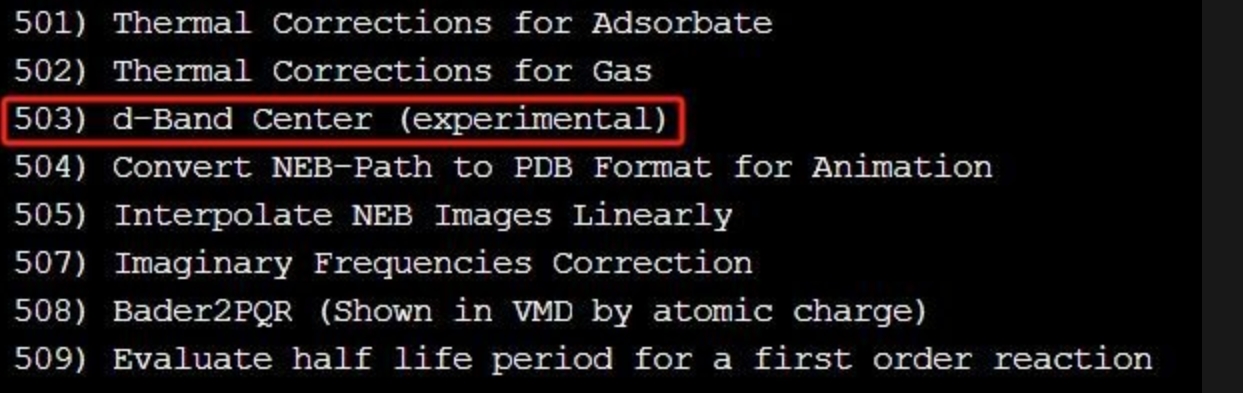
输出文件是 PDOS_SUM.dat, 导出文件用 Origin 打开作图。合并 d-p 轨道的 PDOS 以 MoCrN6 体系吸附 N2 前的投影态密度图为例。吸附 N2 后体系的 PDOS 信息我们可以进行仿照训练。同时运行 vaspkit-5-503, 我们可以得到体系的 d 带中心信息。
我们以 TiMoN6 吸附 N2 为例, AB 代表整个体系的结构, A 代表 TiMoN6, B 代表 N2,分别对 AB、A 和 B 进行静态计算,得到 AB、A 和 B 的电荷密度( 计算输入文件包INCAR、KPOINTS、POSCAR、POTCAR 和 CHGCAR,注意除了 POSCAR 和 POTCAR 不一样, 其他的文件都一样)。
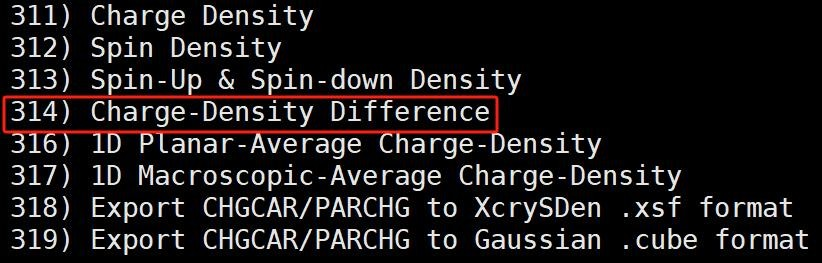
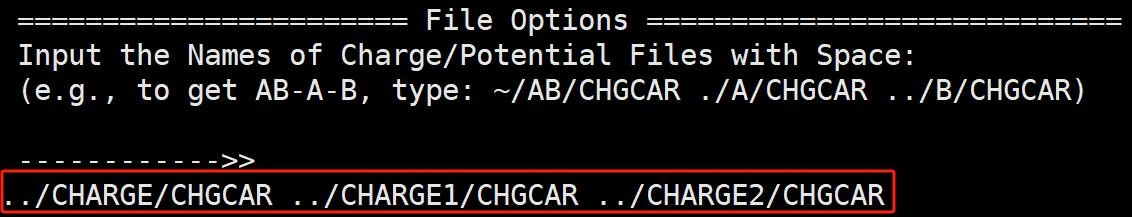
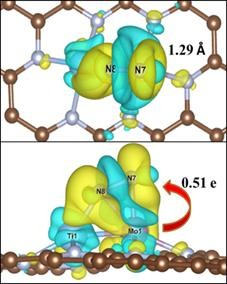
计算完成后我们调用 vaspkit-314 命令, 根据提示依次输入 AB 体系、片段 A 和片段 B 的 CHGCAR 文件所在的位置,就可以得到 CHGDIFF.vasp 文件。将该文件导出到 VESTA 软件中, 即可观察到 TiMoN6 吸附 N2 的差分电荷密度图。
对吸附 N2 后的体系进行结构优化后, 调用 vaspkit-402-1-2-输入需要固定的原子的高度信息,然后调用 vaspkit-501 命令,我们可以得到被吸附物质对于体系吉布斯自由能的零点能校正ΔEZPE 和熵的贡献 TΔS, 带入公式 ΔG = ΔE + ΔEZPE − TΔS, 我们就可以计算反应过程每个中间体的吉布斯自由能, 绘制能量台阶图, 从而确定催化反应的决速步骤。
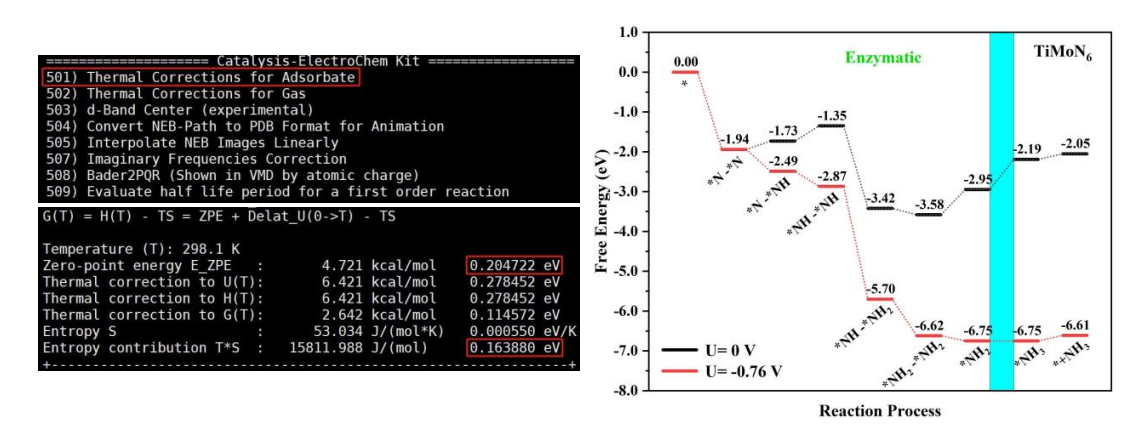
以上, 我们通过电催化氮还原反应实例详解了如何在超算互联网使用 VASPKIT v1.2.5 版本进行第一性原理计算, 通过 vaspkit 模块生成输入文件并对计算结果进行后处理,借助 VESTA 可视化软件以及 Origin 作图软件,得到直观的图像,来分析体系的结构和电子信息。
希望本篇最佳实践为您提供一些 VASPKIT 的实战计算技巧。