高性能计算服务
>
>
VMD(Visual Molecular Dynamics)是一款广泛使用的分子可视化软件,能够帮助研究人员直观地观察和分析复杂的生物分子结构。其强大的可视化功能使得用户能够轻松识别潜在的结构问题,包括手性错误。
AutoIMD是集成在VMD中一个插件,用于自动化地设置和启动NAMD(一种分子动力学模拟软件)模拟。
本文将介绍如何利用VMD、AutoIMD和NAMD,可视化地识别和修正生物分子结构中的手性错误。
手性(Chirality)是指一个物体不能与其镜像完全重叠的现象。在生物分子如蛋白质和核酸中,通常指一个分子不能通过简单的旋转或翻转变成其镜像异构体。例如,大多数生物体内的氨基酸以L-构型存在,而糖则以D-构型存在,这种选择性是生物体识别和相互作用的基础,它影响着分子的识别、反应性和功能。手性的研究和应用正在不断扩展,从药物开发到新材料的设计,手性都扮演着关键角色。
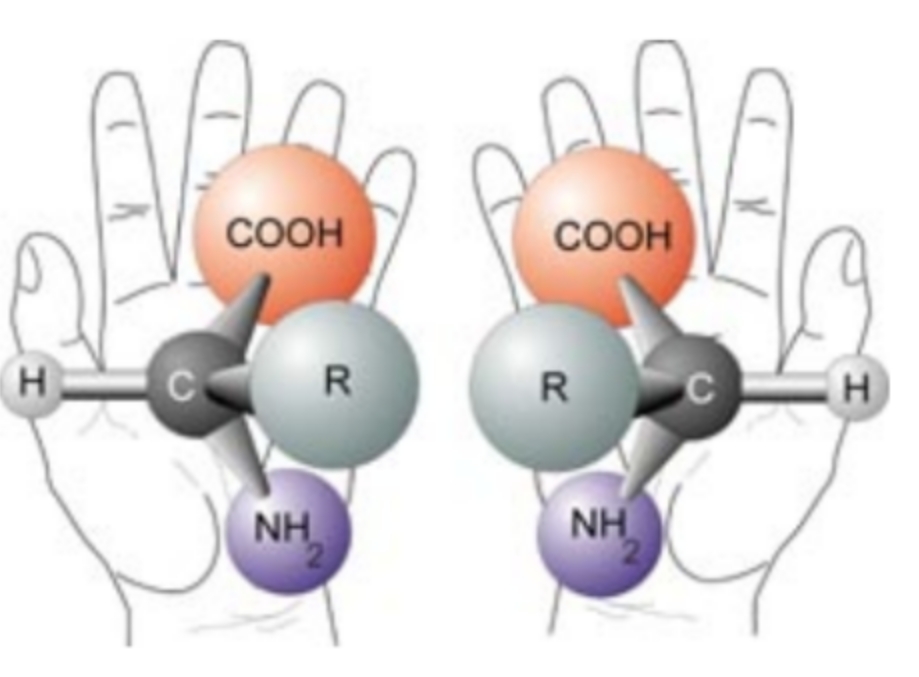
VMD中的手性检查功能允许用户识别和纠正生物分子中的手性错误,这对于维持分子的生物活性和进行准确的结构模拟至关重要。通过使用chirality功能(插件),用户可以检测不寻常的手性中心、可视化错误,并进行必要的结构优化。此外,VMD还能生成用于分子动力学模拟中的额外约束,以保持手性中心的正确构型,确保模拟结果的准确性。
接下来,我们将以VMD官方测试算例为例(该算例人为加入了一些手性错误),基于VMD图形界面演示检查、修正手性错误的整个过程。
本文实验使用超算互联网提供的VMD 1.9.3版本软件,支持图形和命令行两种使用方式。您可通过以下商品链接直接购买使用VMD软件:
算例链接:
https://www.ks.uiuc.edu/Training/Tutorials/science/structurecheck/structurecheck-tutorial-files.zip
在使用VMD进行手性结构检查时,同时需要 PSF(Protein Structure File)和 PDB(Protein Data Bank)文件。PDB 文件通常用于描述生物大分子(如蛋白质和核酸)的结构,而PSF 文件提供了分子模型的详细结构和力场参数。在手性结构检查中,VMD 需要了解分子的几何结构(来自PDB文件)以及分子的拓扑信息(来自PSF文件)。官方算例提供了chir testcase.psf和chir testcase.pdb文件,我们需要同时将二者导入VMD。
由于后续需要使用NAMD,为了操作方便,我们通过命令行的方式打开VMD图形界面。使用步骤和关键命令如下:
#加载VMD环境变量
source /PATH/TO/YOUR/VMD/1.9.3-none/scripts/env.sh
#设置显示端口号,需要打开Linux图形桌面并查看display号
export DISPLAY=your_display_port
#打开vmd gui
vmd操作完成后,则会在linux图形桌面处显示打开的VMD主界面和Display窗口。
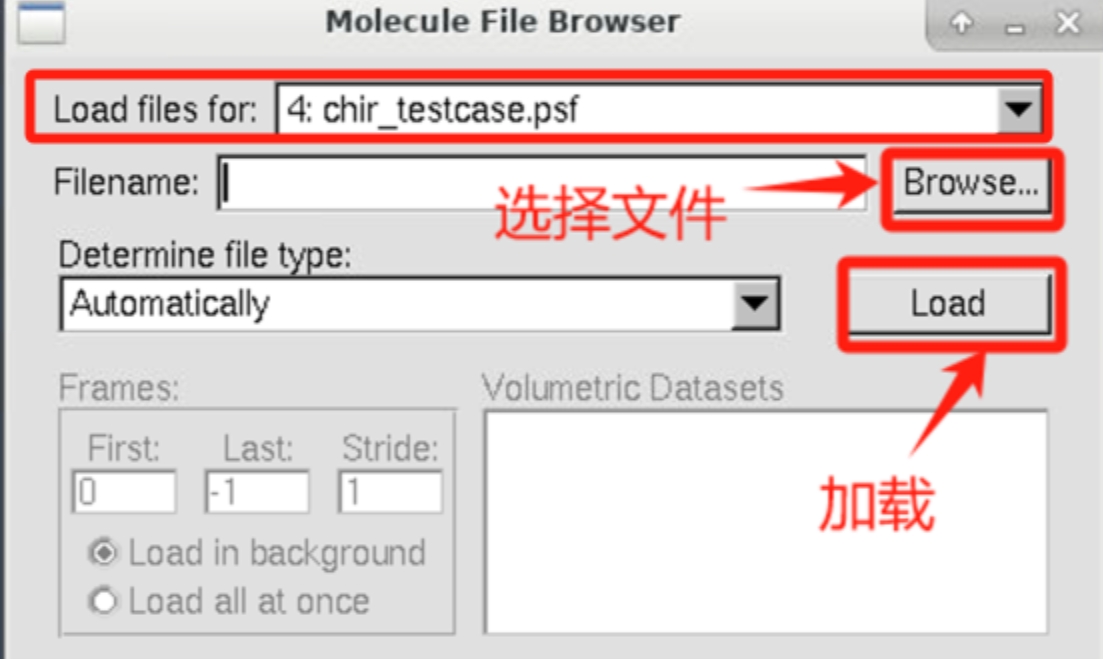
在“VMD main”主窗口选择“File”—>“New Molecule...”,在弹出的文件选择器中,分别加载psf和pdb文件。我们建议先加载psf文件,因为 PDB 文件中的坐标需要与 PSF 文件中的拓扑信息相匹配。如果先加载 PDB 文件,VMD 可能无法正确解释原子之间的连接关系,因为这些信息包含在 PSF 文件中。加载了 PSF 文件之后,VMD 会根据 PSF 文件中的拓扑信息来构建分子模型,然后再用 PDB 文件中的坐标信息来定位每个原子。
加载完成后,我们可以看到VMD Display窗口已经有了结构显示。这里顺便介绍下VMD视图的快捷键:“R”按键后,我们可以通过鼠标旋转视图;“T”按键后,可以拖拽调整视图;“S”键可以放大和缩小;“Y”键则会自动保持旋转。
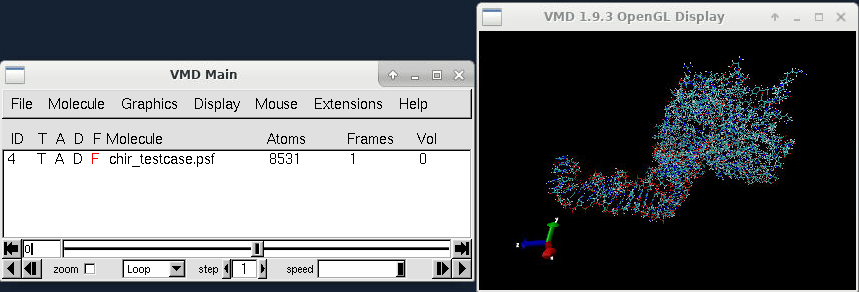
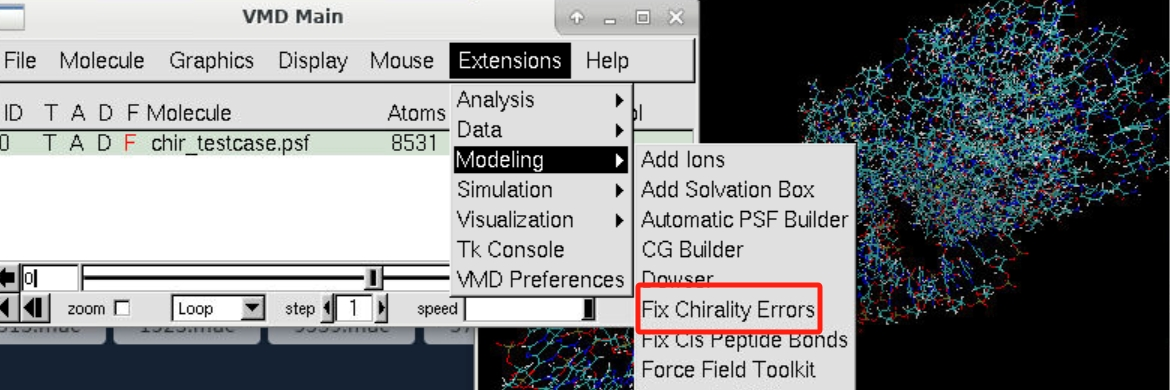
导入完分子结构后,接下来我们需要使用VMD的chirality功能自动检测结构中不合理的结构。在主窗口中选择我们导入的分子,然后在功能菜单中选择“Extensions”—>“Modeling”-->“Fix Chirality Errors”,即可弹出“Chirality”功能选项卡,具体操作可参考下图。
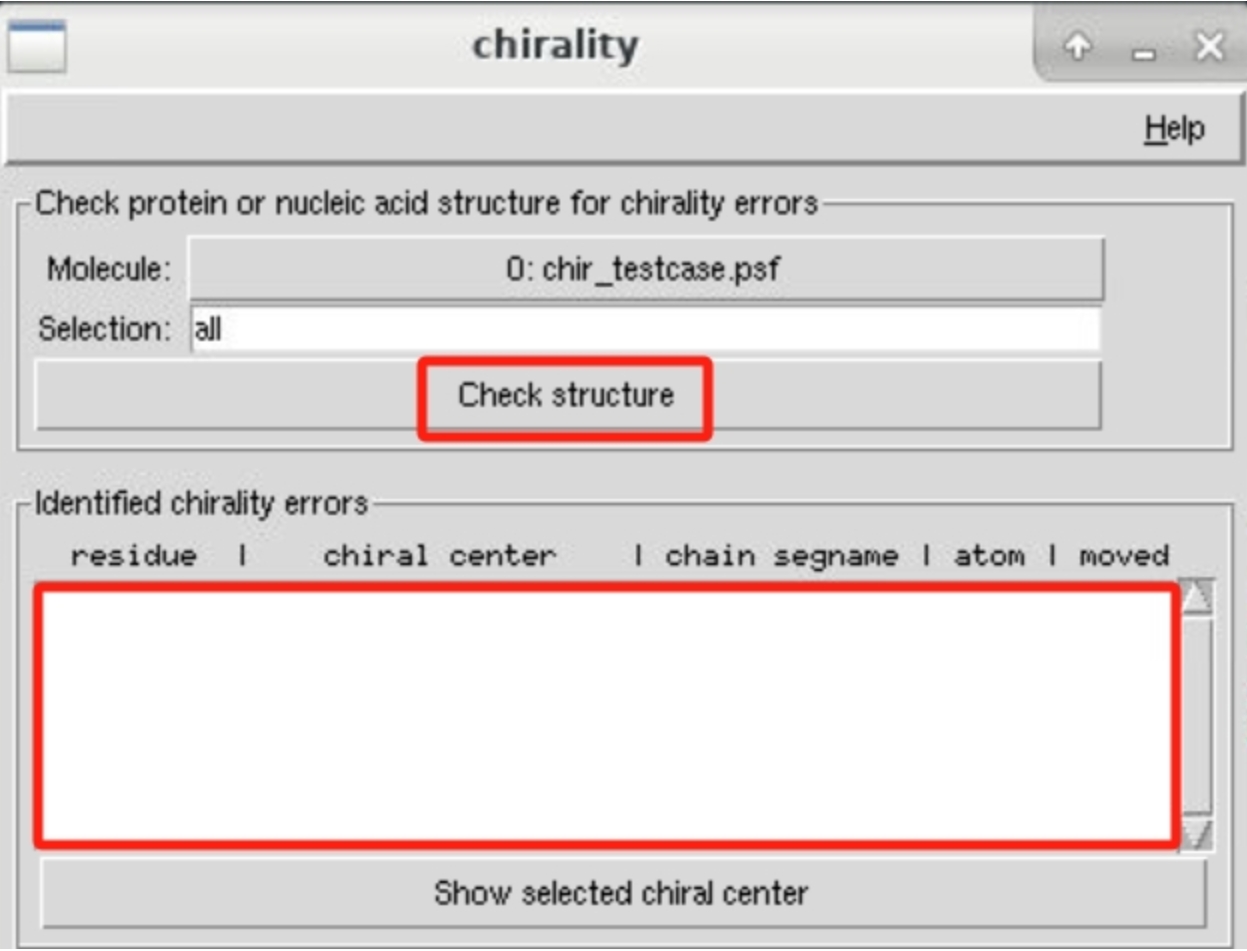
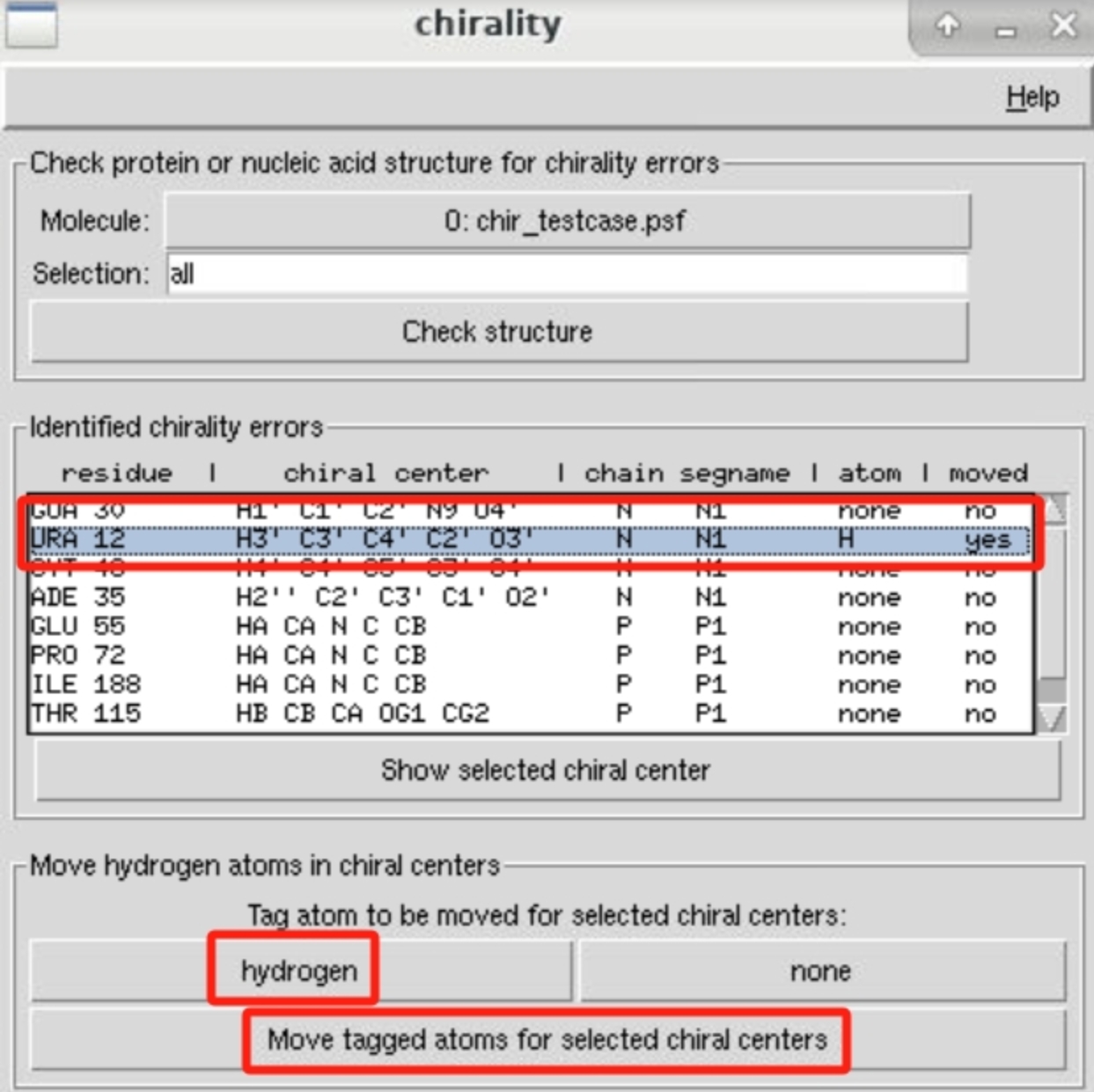
chirality主界面如下图所示。“Selection”字段允许用户输入特定的选择表达式,以定义要检查的分子部分,通过选择特定的原子或残基等,用户可以更精确地进行手性检查。这里我们默认选择“all”,检查整个分子。设置完成后,点击“Check structure”,则会自动检测手性错误,并会将检测到的手性错误列在“Identified chirality errors”文本框中。
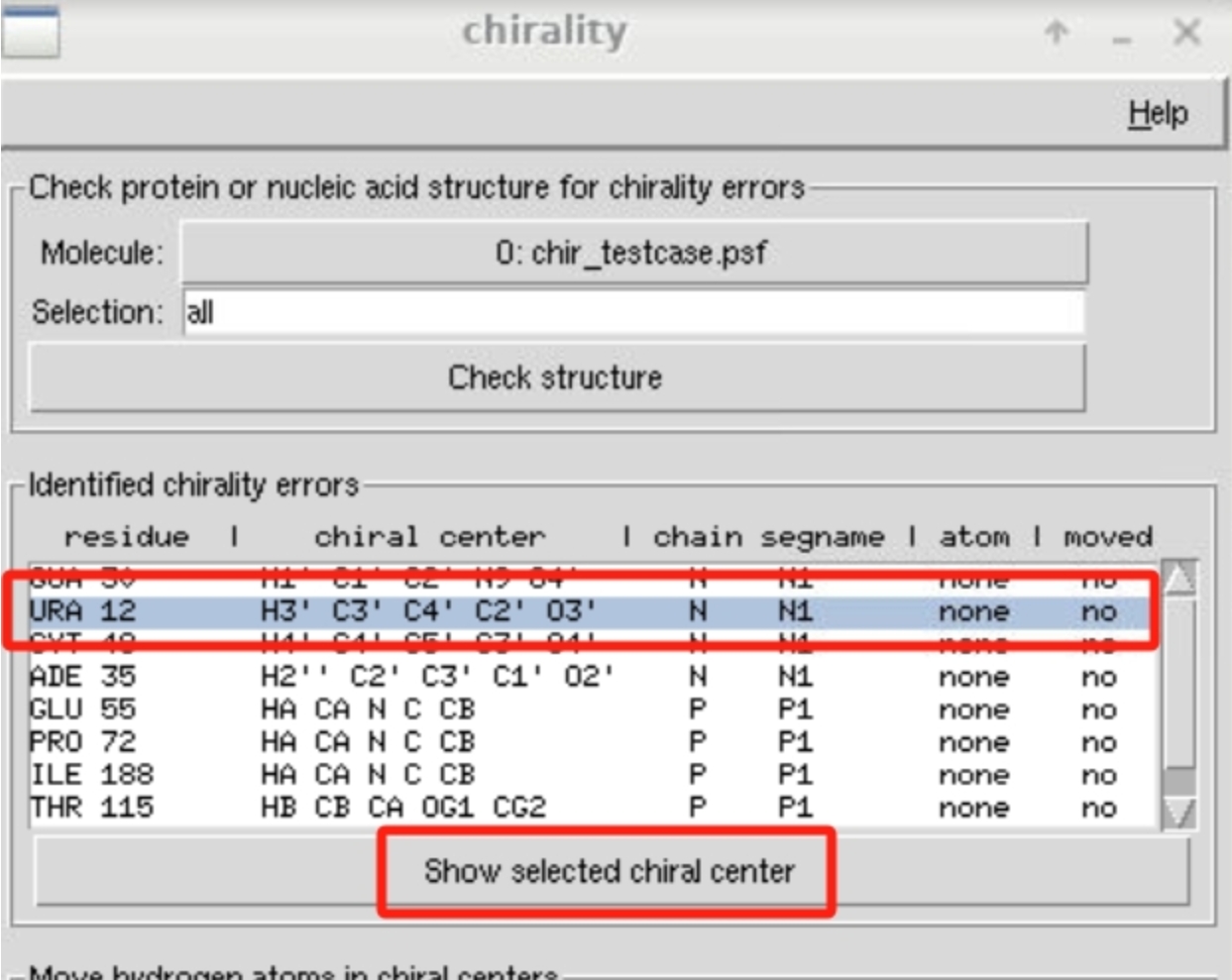
实验算例检测到的手性错误如下图所示。VMD中的手性分子检查功能主要用于确保手性中心的正确性、消除几何不合理性。主要涉及到两个操作:
1)移动氢原子:在手性分子检查中,用户可以通过移动氢原子来调整分子的构型。这是因为氢原子通常在分子中占据较小的空间,移动它们可以有效地改善分子的几何结构。
2)优化结构:在调整氢原子位置后,执行能量最小化或者平衡操作可以进一步优化分子的构型,确保所有原子处于合理的位置,降低系统的总能量、让系统在给定的温度和压力条件下达到热力学平衡。
我们先操作移动氢原子,移动后的进一步结构优化将会使用AutoIMD插件,我们放在后续章节演示。
我们在“Identified chirality errors”列表中选择想要修改的手性错误。点击“Show selected chiral center”,可以在display中看到详细的几何结构,进一步确认我们是否想要通过移动氢原子改变当前结构并进行后续操作。
点击“hydrogen”来标记氢原子,VMD会将与选定的手性中心相关的氢原子标记为待移动的原子。点击“Move tagged atoms for selected chiral centers”按钮,VMD 将根据用户的选择移动标记的氢原子,以修正手性错误。
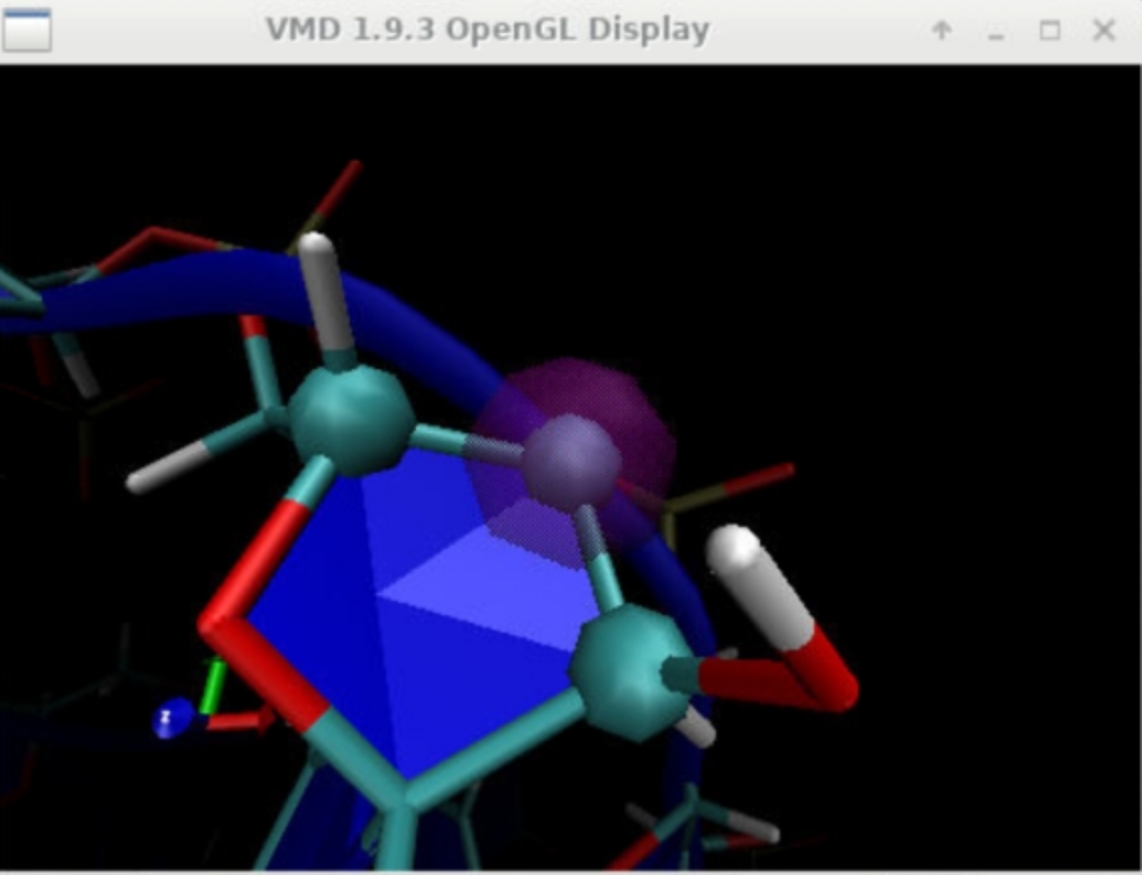
在VMD的chirality插件中,移动氢原子以修正手性中心的构型是基于分子的立体化学规则和几何考虑的。移动完成后,VMD 会更新分子的三维结构,下面两张图从两个角度展示了移动后的三维结构。
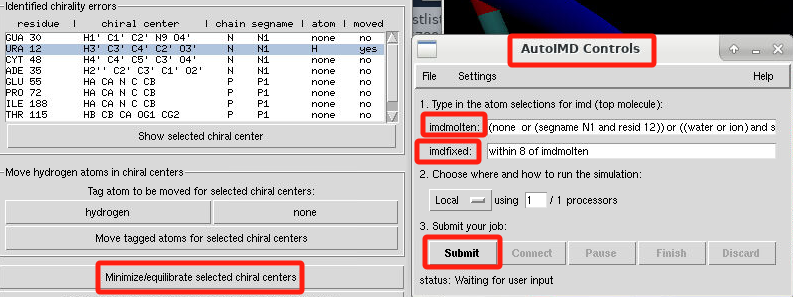
由于只是简单的移动氢原子会产生分子的非物理几何形状,有必要进行优化结构,VMD的手性插件chirality使用AutoIMD来可视化执行优化步骤。由于AutoIMD会调用NAMD执行分子动力学模拟,我们需要提前设置好NAMD软件。
NAMD是一个用于大规模并行模拟的分子动力学软件,它能够模拟从纳米尺度到微米尺度的分子系统,进行常规分子动力学模拟、能量最小化、自由能计算等模拟。
超算互联网商城提供了多个NAMD版本,满足您即需即用的要求。本文实验使用NAMD 2.14版本软件,只需要在命令行执行source 软件scripts目录下的env.sh即可使用软件。
点击“Minimization/equilibrate selected chiral centers”,我们可以打开AutoIMD Controls窗口,选择执行能量最小化或者平衡操作。使用AutoIMD时,可以将模拟系统分为三个不同的区域:熔化区、固定区和排除区。熔化区表示原子自由移动的区域,通过imdmolten指定该区域;imdfixed指定被排除的区域,在模拟执行时会被忽略。
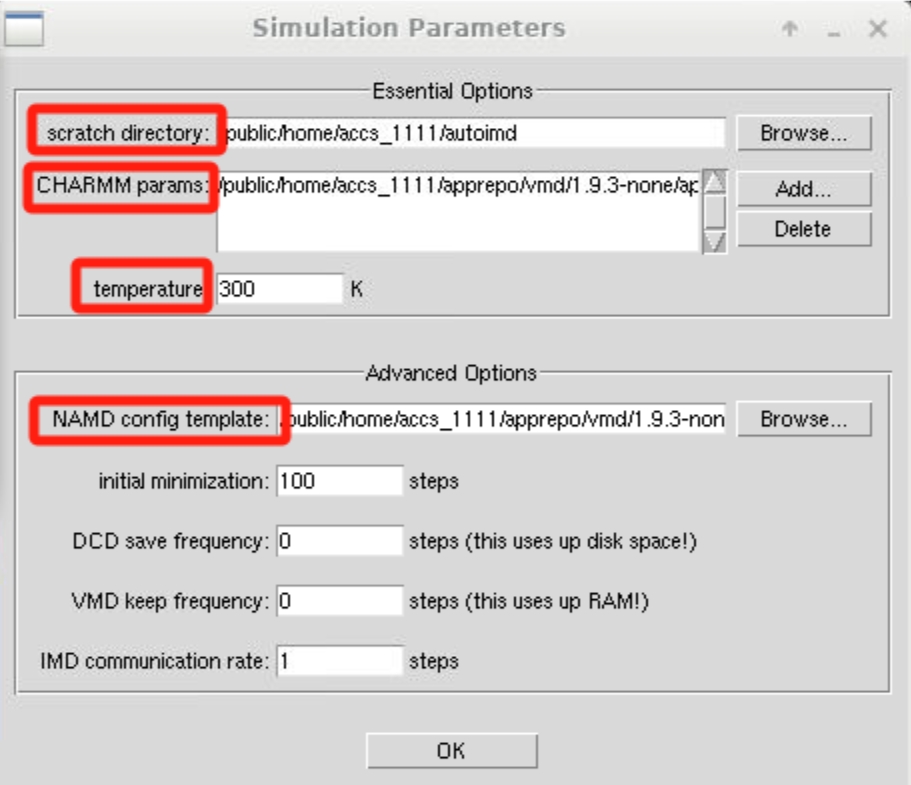
“Settings”选项允许您选择设置模拟的参数,比如指定临时工作目录、设置自己的立场参数文件、指定温度和指定NAMD配置文件。这里我们使用默认的CHARMM立场文件和vmd自带的NAMD模板配置文件演示自动启动NAMD进行能量最小化模拟。
设置完成后,可以在AutoIMD主界面点击“Submit”来提交模拟,后台自动完成一些初始化后,“Connect”按钮变成可点击状态,用来执行启动NAMD软件,自动执行可视化模拟。第一次使用时,需要手动设置NAMD的可执行二进制文件位置,以便AutoIMD能够顺利找到NAMD并开启连接,若连接失败,请确认配置好NAMD的环境变量后多试几次。连接成功后,就可以实时进行优化模拟了。
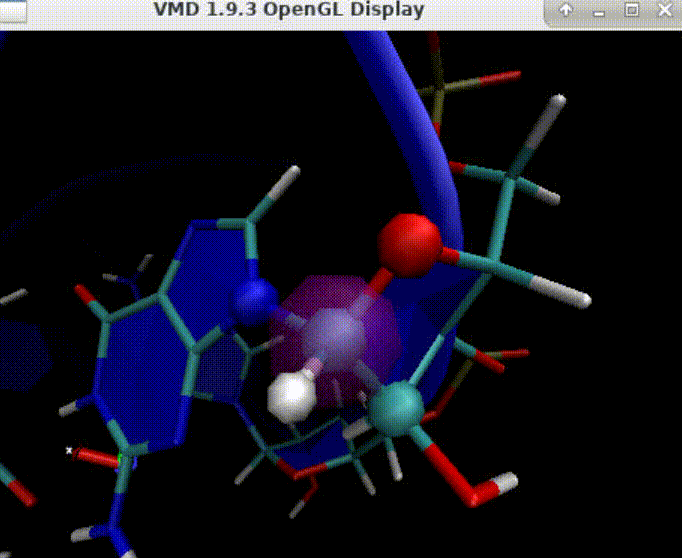
您可以点击“Pause”按钮暂停运行,或者检视到结构满足需求了,可点击“Finish”提前结束NAMD运行,“Discard”按钮也会停止NAMD模拟,但是不更新坐标,体系会回到初始状态,如果出问题,这个按钮可以返回初始状态。
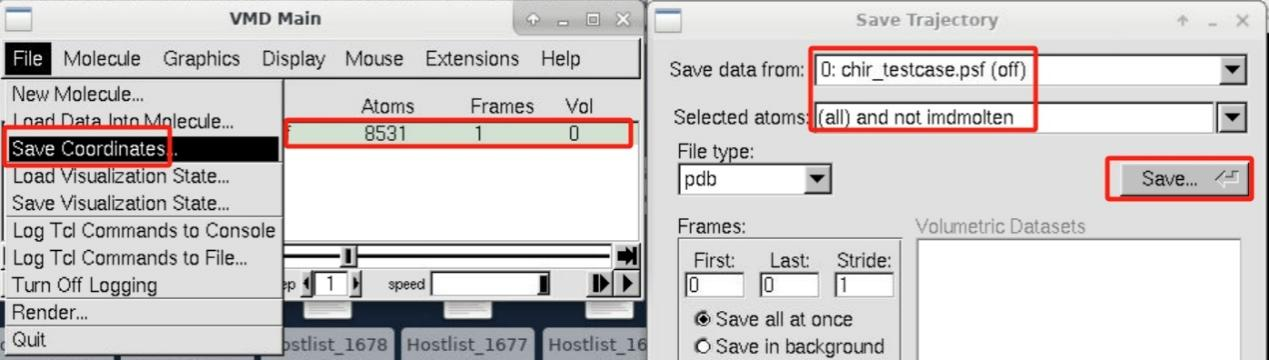
当手性错误修正完成后,我们需要保存修正后的结构。在VMD主窗口中,选择“File”-->“Save Coordinates...”在弹出的保存窗口中,“Selected atoms”选择“all”,并选择想要保存的文件类型后,点击“Save”,即可将新的结构保存。
在本篇实践中,我们详细探讨了如何运用VMD、AutoIMD和NAMD这三款工具,对生物分子结构中的手性错误进行可视化识别与修正。通过这些方法,研究人员能够更准确地把握分子结构,确保生物信息学研究和实验结果的可靠性。
希望本篇最佳实践为您提供一些指导和建议。