高性能计算服务
>
软件使用案例
>
科学计算
>
Gromacs
本文主要介绍如何在超算互联网商城使用Gromacs软件。
在商城首页输入软件名搜索软件,点击需要使用的软件卡片进入软件商品界面,选择软件版本以及资源配置区域,阅读并同意《服务协议》,点击“立即使用”等待配置完成可以使用该软件;已经使用过的软件会在规格的右上角有一个绿色的“已使用”标识,点击“去使用”可直接使用该软件。
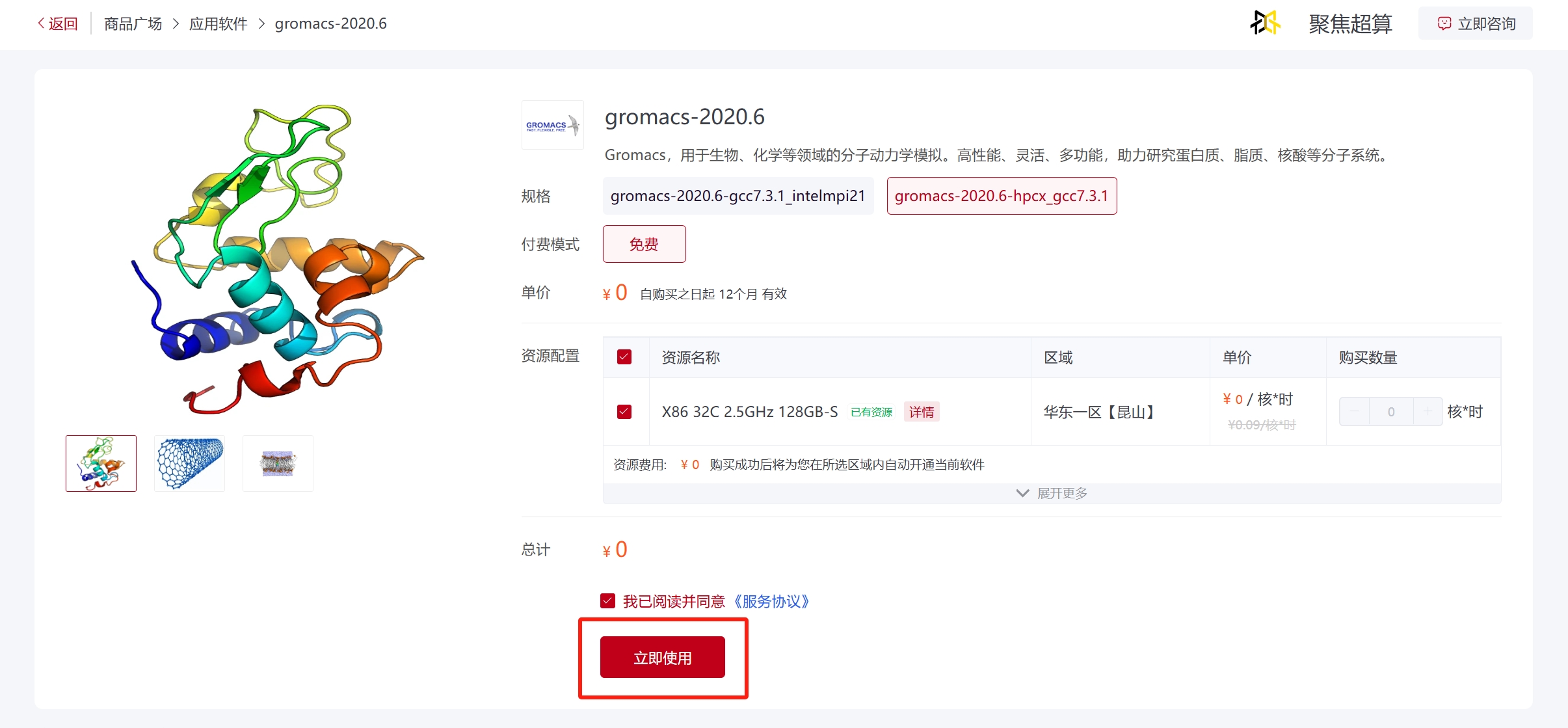
已经使用过的软件亦可以选择其他可用区域开通软件,或点击“已开通”后面的提交作业方式直接使用。
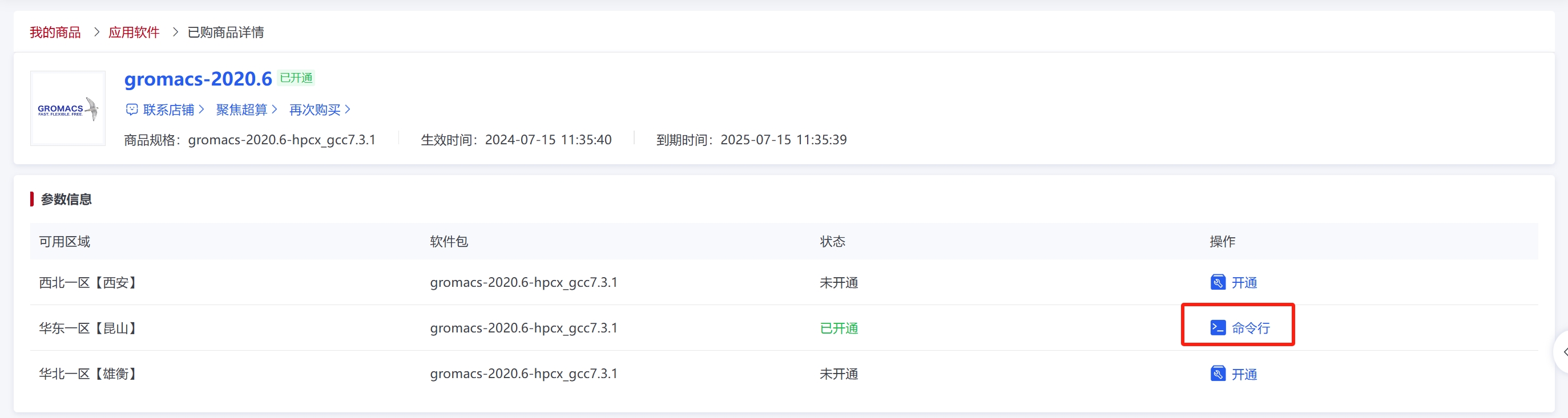
点击“命令行”图标后进入eshell界面,从商城的“命令行”进入终端,终端会显示商品自带的软件应用版本、环境变量和启动路径信息。由启动路径可以得到软件部署的位置。(注:实际使用中请根据下方红框内提供的算例示例进入对应路径。)
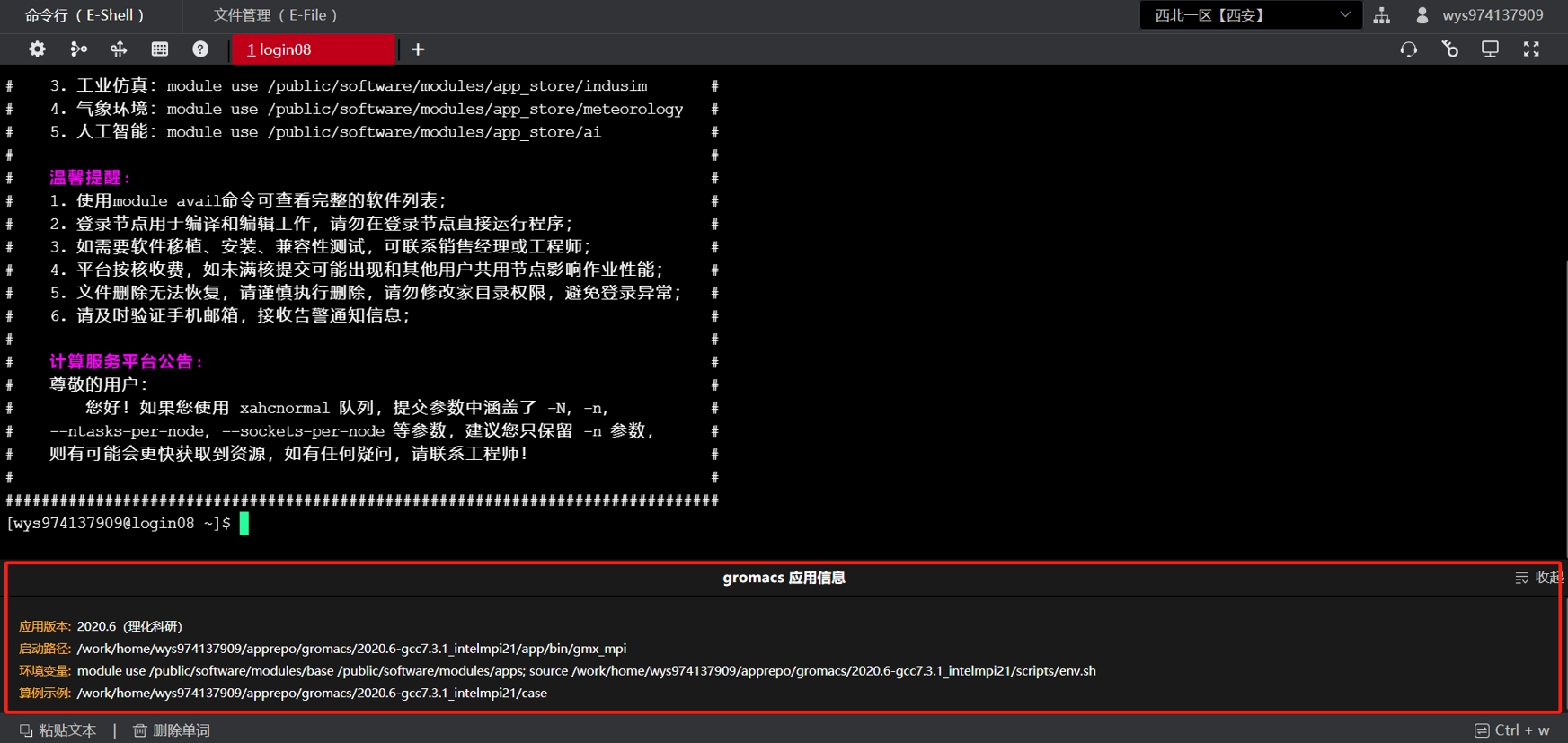
本产品软件安装目录为 ~/apprepo/gromacs/2020.6-gcc7.3.1_intelmpi21/,包含app(除快速安装软件外)、case、install.log、scripts四个子目录,其中app包含应用软件主体和依赖库;case包含相关运行算例、作业日志、作业提交脚本等信息;install.log为GROMACS安装日志,scripts为软件环境变量。目录结构如下:
.
├── app # 应用软件主体和依赖库
│ ├── . . .
├── case # 可运行算例、脚本
│ ├── gromacs.slurm # slurm脚本文件
│ ├── slurm-7016471.out # 运行日志
│ └── . . .
├── install.log # GROMACS安装日志
└── scripts # 应用环境变量
├── env.sh
└── post.sh
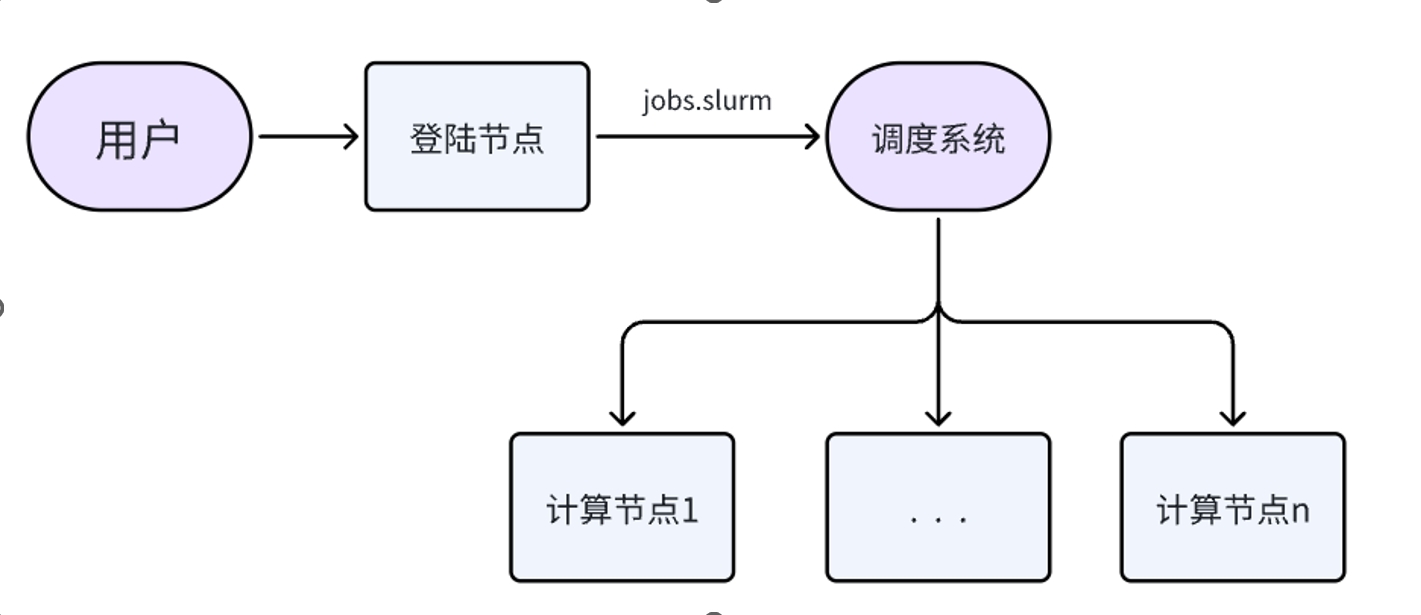
资源调用逻辑如下:用户在登录节点执行脚本,脚本将自动申请计算资源,并在该计算资源上运行计算指令,并将计算结果实时同步到当前目录下。
我们已经为您提供完整的使用脚本:
(1) 执行cd ~/apprepo/gromacs/2020.6-gcc7.3.1_intelmpi21/进入软件部署目录,在目录下的case文件夹中有软件对应的提交脚本。

(2) 输入whichpartition可以查询可用队列,复制选中合适的队列名称,这里选择了“xahcnormal”,并打开脚本gromacs.slurm进行脚本文件的修改。
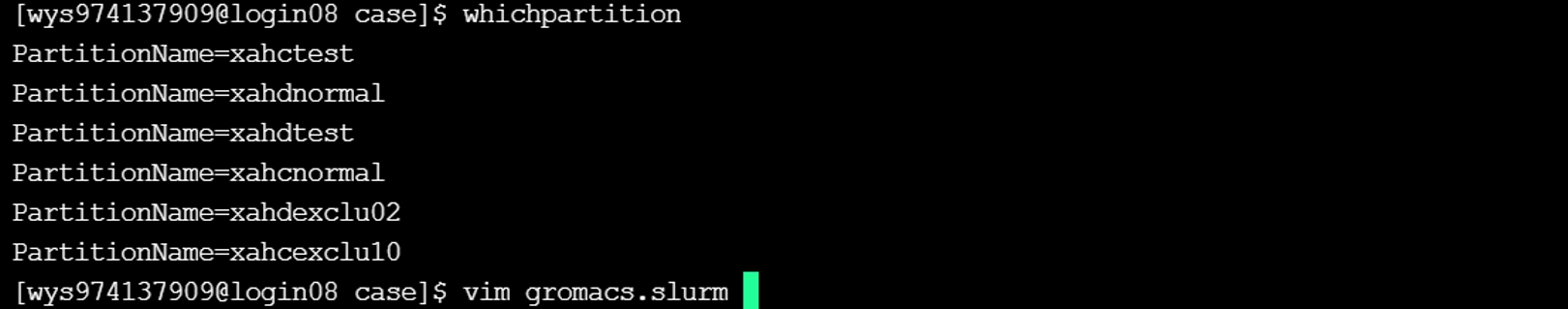
(3) 替换脚本gromacs.slurm中的队列名称为复制好的队列名称,并根据自己的需求更改脚本的配置信息。
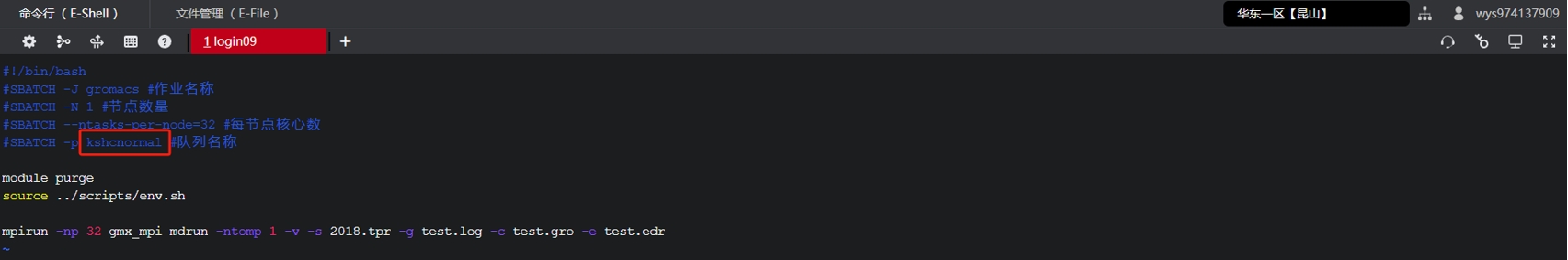
(4) 如果您需要进行其他修改可参考如下命令:
| 参数 | 含义 |
|---|---|
| -J gromacs | 指定作业名为gromacs |
| -p xahcnormal | 指定使用队列为xahcnormal |
| -N 2 | 申请两个计算节点 |
| --ntasks-per-node=32 | 每个节点使用32核 |
注意:
1) -J、-p、-N、--ntasks-per-node=32为slurm调度参数,更多调度命令可使用sbatch --help命令查看;
2) slurm脚本中的命令依次执行,提交作业时需把脚本和输入文件放在相同文件夹,并在该文件夹下提交作业;
3) 该示例以仅供参考,其他中心资源请根据实际对应修改队列、核数和软件环境等;
4) 上述描述为命令行提交作业方式,除此之外还有模板提交和图形提交两种方式(需软件自身支持),具体见“其他作业提交方式”。
(1) 确认环境变量、修改好脚本后保存退出,sbatch gromacs.slurm 提交作业,并通过squeue 查看作业的运行状态。
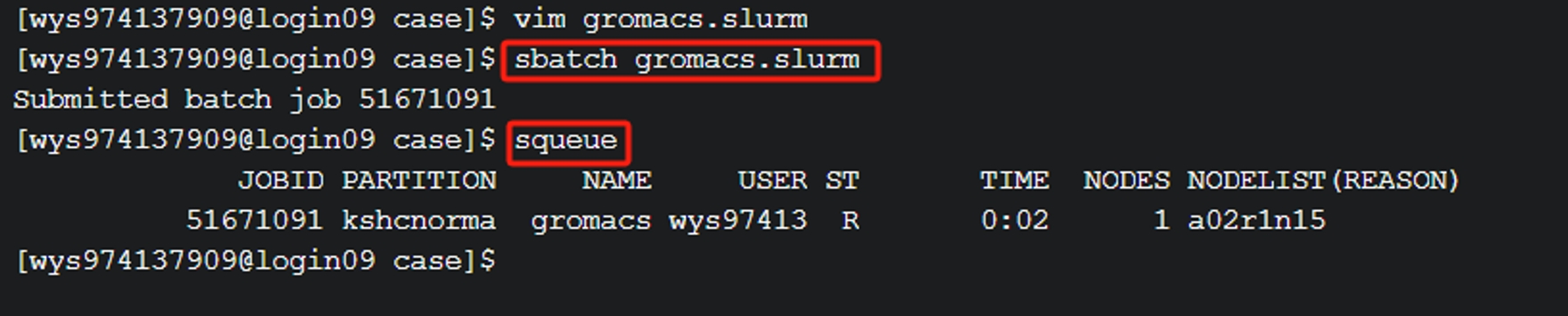
(2) 任务完成后作业会自动结束,并在当前目录生成日志文件slurm-作业号.out或使用tail -f slurm-作业号.out实时查看。

(3) 打开slurm-11087910.out查看作业的日志输出以及检查是否正常结束。
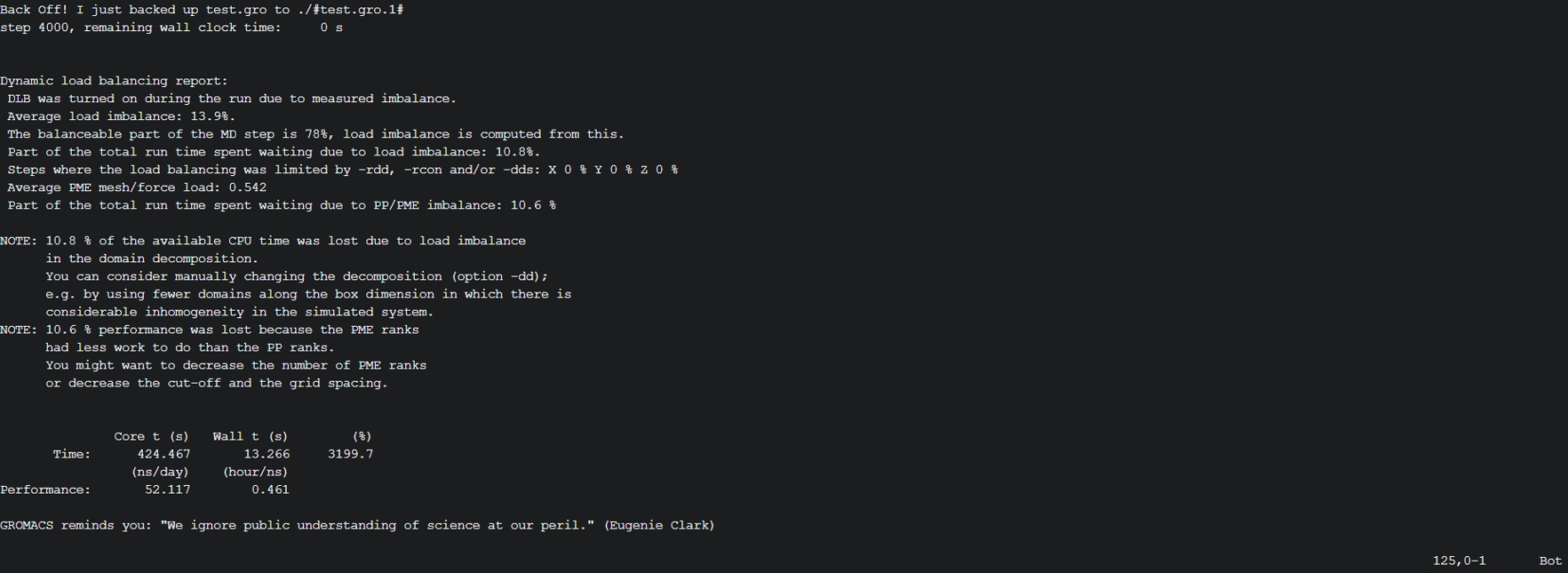
(4) 另外可参考如下常用指令进行作业管理:
| 命令 | 含义 | 简单示例 |
|---|---|---|
| sbatch | 批量提交作业命令,后面跟脚本文件 | sbatch xxx.sh |
| squeue | 查看目前提交作业的信息 | squeue(可显示作业号、作业状态等) |
| salloc | 占用空闲计算资源命令 | salloc -p kshctest -N 1 -n 32 |
| scontrol | 查看正在计算作业信息 | scontrol show job jobid |
| scancel | 取消作业 | scancel jobid |
| sacct | 查看历史作业 | sacct -j jobid -X -o elapsed,state,nodelist |
注意:请不要在登录节点(login)上直接运行作业计算(编译等日常操作除外),以免影响您的作业正常运行。
(1)gromacs力场文件放到哪里?
解决思路:自行准备的gromacs力场文件可以拷贝至top目录下,如:~/apprepo/gromacs/2022.2-hpcx_gcc7.3.1/app/share/gromacs/top/gromos54a7_atb.ff。
(2)gromacs-dcu/gpu报错:PME GPU does not support PME decomposition 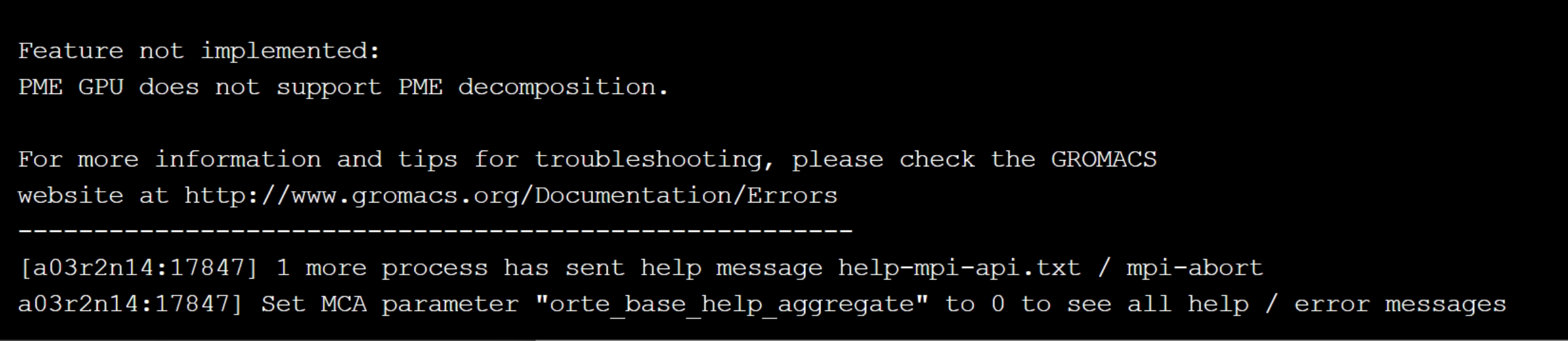
解决思路:脚本中写入pme方法,需要单独指定-npme 1。如:>>> mpirun --allow-run-as-root -np 4 gmx_mpi mdrun -ntomp 7 -nb gpu -pme gpu -npme 1 -v -noconfout -deffnm nvt -nsteps 1000000
(3)gromacs报错:There is no domain decomposition for 120 ranks that is compatible with the given box and a minimum cell size of 0.4975 nm
解决思路:可以通过修改gromacs运行核数、加-dds、加-rdd(0-1.0)参数来尝试修正解决,如: 1)mpirun -np 16 gmx_mpi mdrun -s umbrella7.tpr -deffnm umbrella7 -pf pullf-umbrella7.xvg -px pullx-umbrella7.xvg -v -dds 0.9 2)mpirun -np 16 gmx_mpi mdrun -s umbrella7.tpr -deffnm umbrella7 -pf pullf-umbrella7.xvg -px pullx-umbrella7.xvg -v -dds 0.5
(4)gromacs报错:setting the number of the thread-MPI ranks is only supported with thread-MPI 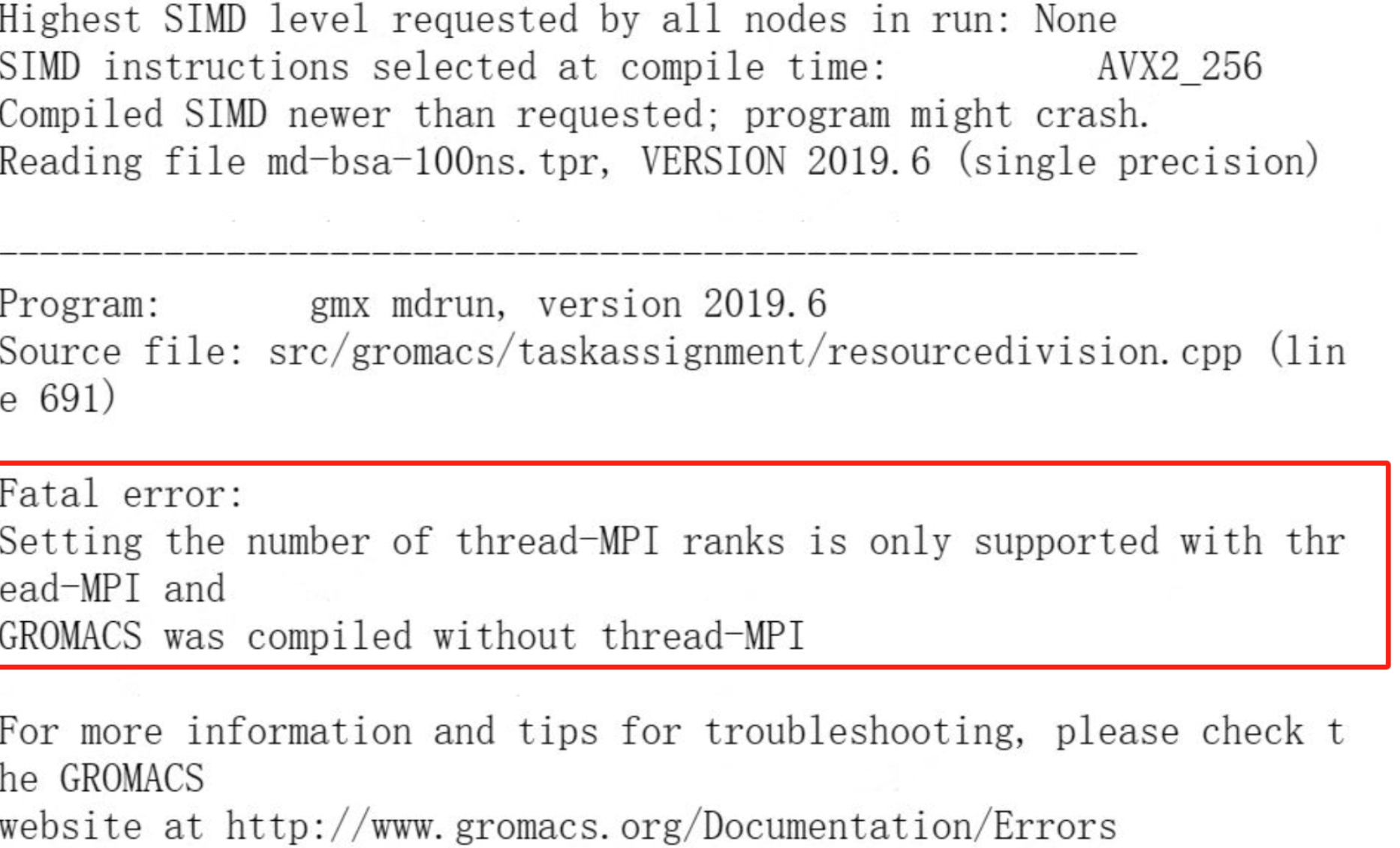
解决思路:mpi版本gromacs无需使用-ntmpi参数,使用mpirun即可。如:gmx_mpi mdrun -pin on -ntmpi 4 -ntomp 8 -nb gpu -bonded gpu -pme gpu -npme 1 -v -deffnm -nsteps 20000应修改为:mpirun -np 4 gmx_mpi mdrun -pin on -ntomp 8 -nb gpu -bonded gpu -pme gpu -npme 1 -v -deffnm -nsteps 20000